Research
Molecular Machines
Molecular Motors
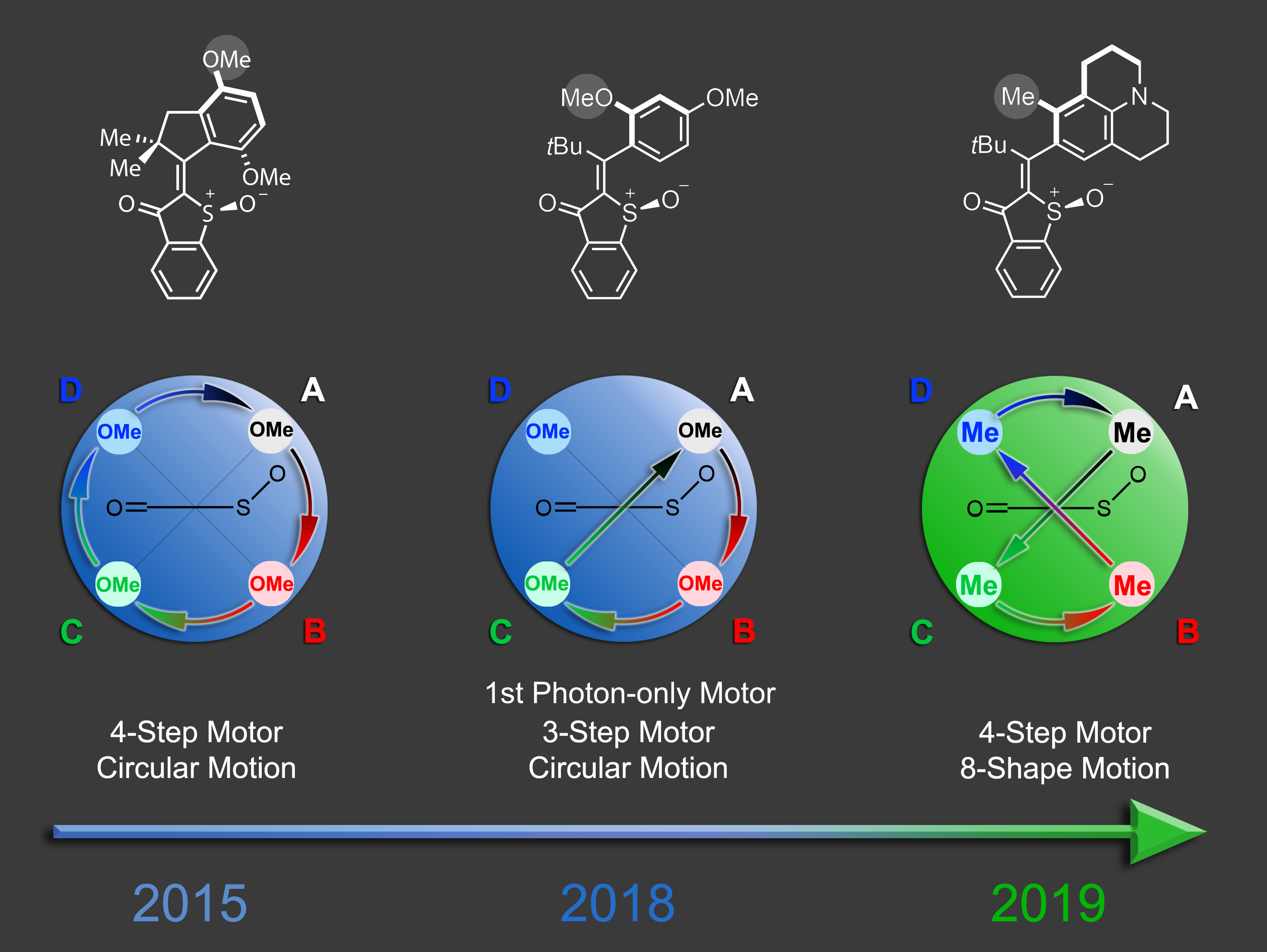
Molecular machines represent the quintessential realization of Feynmans 1959 vision – to fill the “plenty of room at the bottom” with intricate functions. We have contributed to this rapidly evolving field by generating fast and visible light driven molecular motors that are highly promising for applications not tolerant of damaging UV light or high temperatures.1 After developing new synthetic methods for their manufacturing and gaining control over their maximum rotation speeds2 we recently provided a comprehensive mechanistic picture of their mode of action.3 From these studies crucial factors to influence their efficiencies and future design opportunities could be determined.
1. Nat. Commun. 2015, 6, 8406.
2. Angew. Chem. Int. Ed. 2017, 56, 14536.
3. J. Am. Chem. Soc. 2018, 140, 5311.
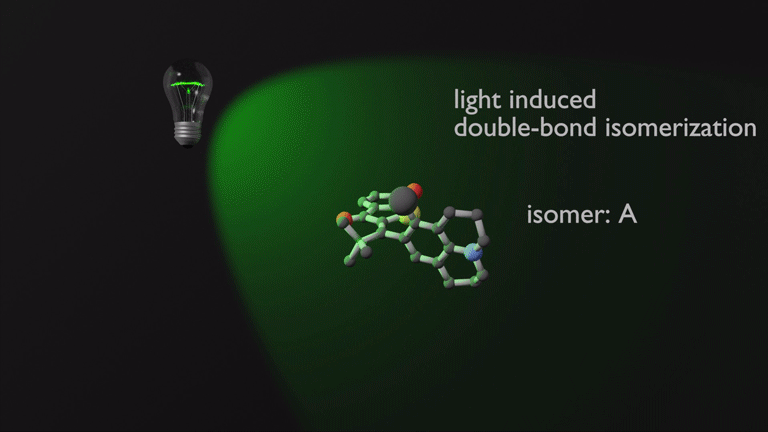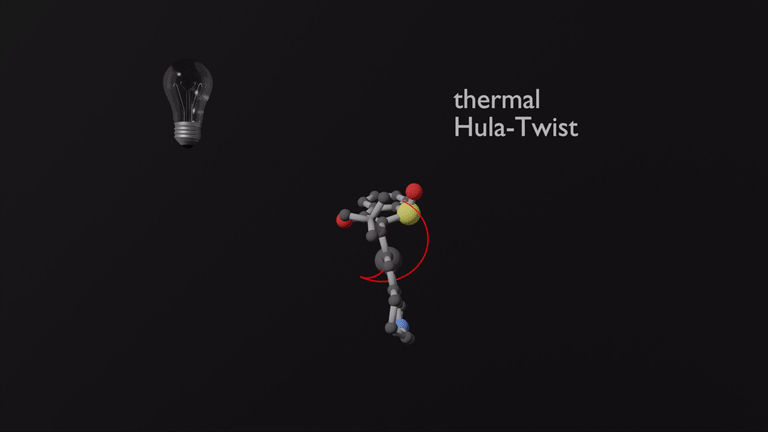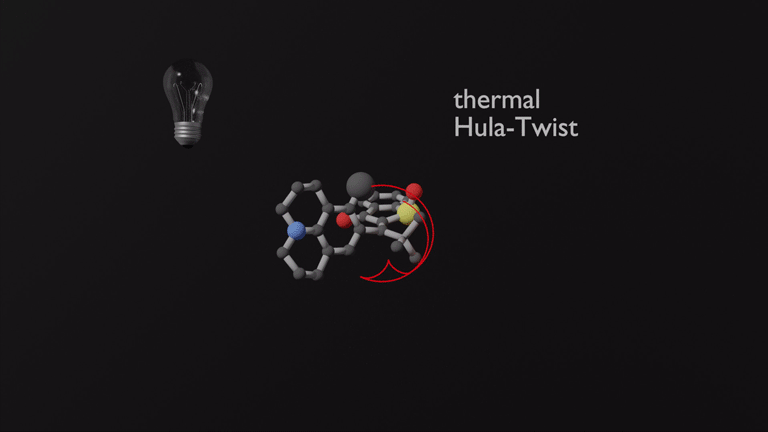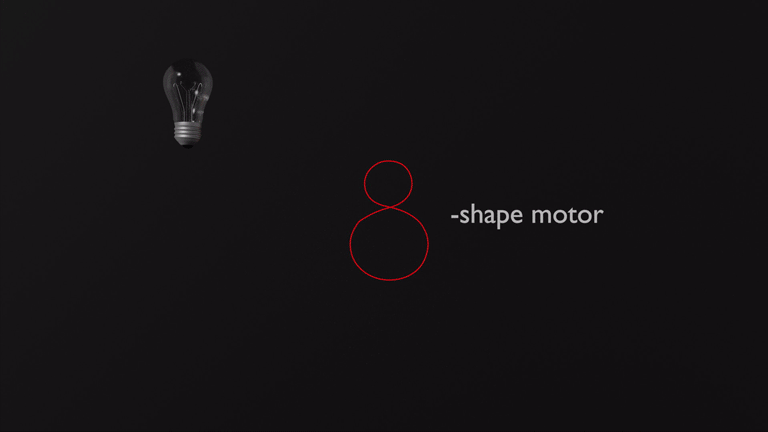
In recent breakthroughs in our laboratory we achieved the development of two entirely new types of molecular motors. A photon-only driven motor works without thermal ratcheting in the ground state and is powered by three consecutive light-induced steps. Because of this unique mechanism a reverse temperature dependency of its efficiency is observed, i.e. this motor becomes faster at lower temperatures instead of slower.4 Our latest motor development enables more complex directional motions than the hitherto possible linear or circular trajectories. Powered by green light the molecular motor undergoes an eight-shape motion, which is fully directional.5 These developments will open up new avenues for molecular machines and their applications in the near future.
4. J. Am. Chem. Soc. 2018, 140, 16442.
5. Nat. Commun. 2019, 10, 4449.
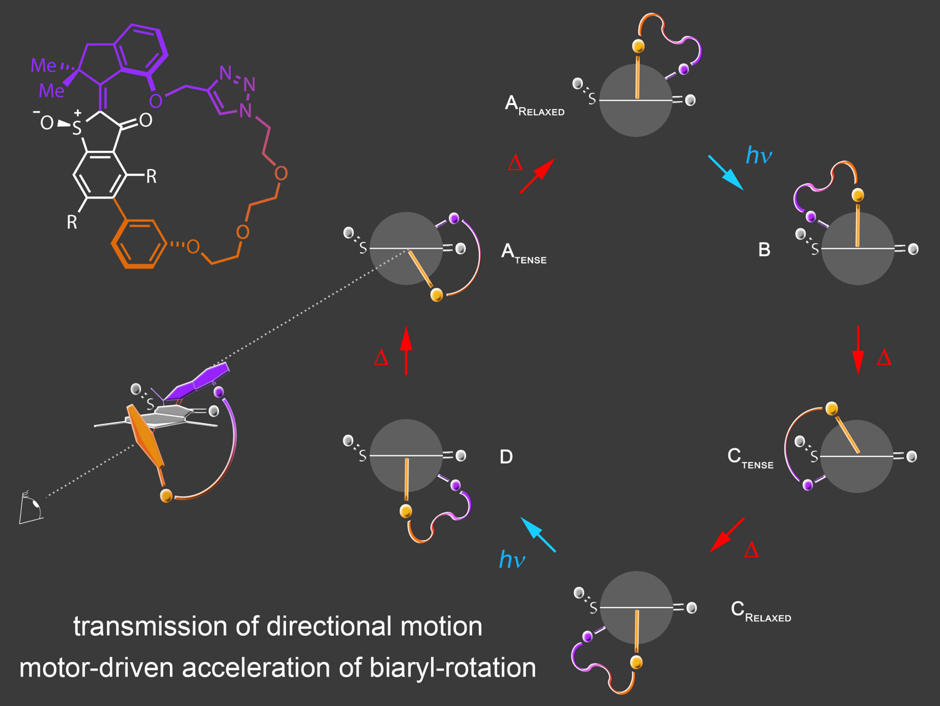
Our current efforts are directed at applications in integrated molecular machines and implementation of our motors into more complex environments. In this regard we have achieved transmission of motor motion unto passive biaryl axes6 in a macrocyclic setup. Subsequently, we were able to use the unidirectional motor rotation to actively accelerate biaryl rotations by several orders of magnitude.7
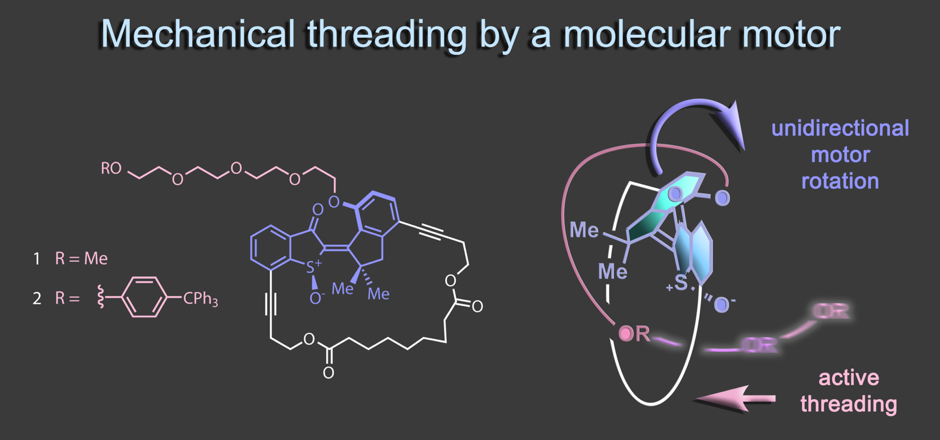
In a recent work the first mechanical threading event powered by a molecular motor has been realized.8 Embedding the motor into a different macrocyclic structure allows us to use the indanone fragment as revolving door during unidirectional rotation. During this rotation an attached tetraethylene glycol chain is actively threaded through the macrocycle in a fully directional way. This concept establishes a first entry point into the uncharted territory of mechanical molecular weaving.
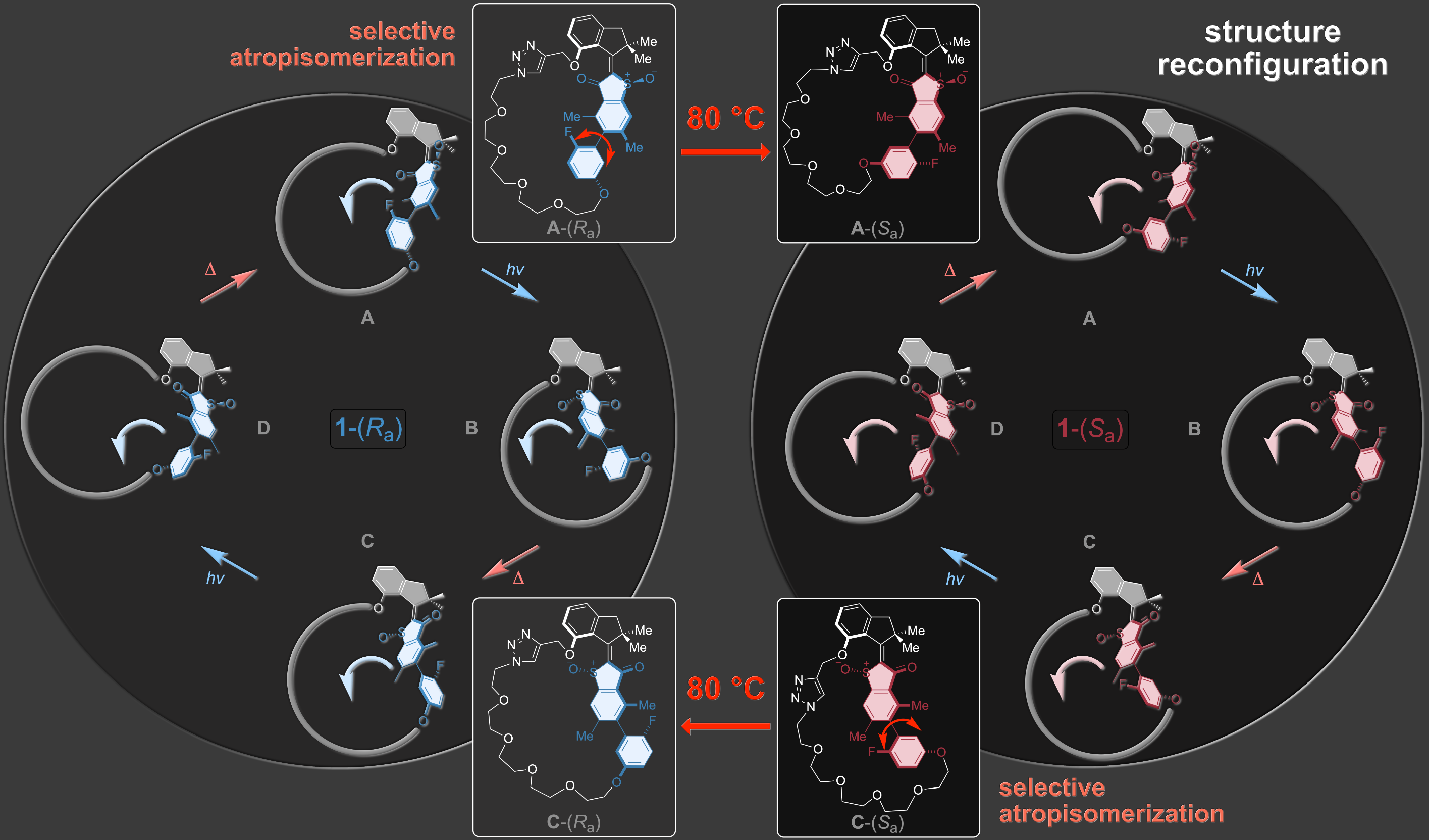
A highly complex reconfiguration of molecular motor rotation can also be elicited within a macrocyclic architecture. A supersized revolving door element rotates within two different larger macrocycles with the same sense of directionality at ambient temperatures. Heating allows to interconvert the two macrocycles reversibly by dissecting the revolving door element allowing to chose within which macrocycle setup unidirectional rotation should occur.9
Further use of macrocyclization allows elucidating the directionality of very fast molecular motors, which cannot be analyzed with physical methods in this regard. Within locked macrocyclic motors all possible directionalities can be dissected and characterized individually by quantum yields. Comparison with the free, non-macrocyclized motor candidate allows establishing unidirectionality and even to obtain information about its degree.10
6. Angew. Chem. Int. Ed. 2018, 130, 11231.
7. Angew. Chem. Int. Ed. 2020, 132, 5730.
8. Angew. Chem. Int. Ed. 2022, 61, e202201882.
9. J. Am. Chem. Soc. 2023, 145, 13081.
10. Nat. Commun. 2023, 14, 4595.
Molecular Gears
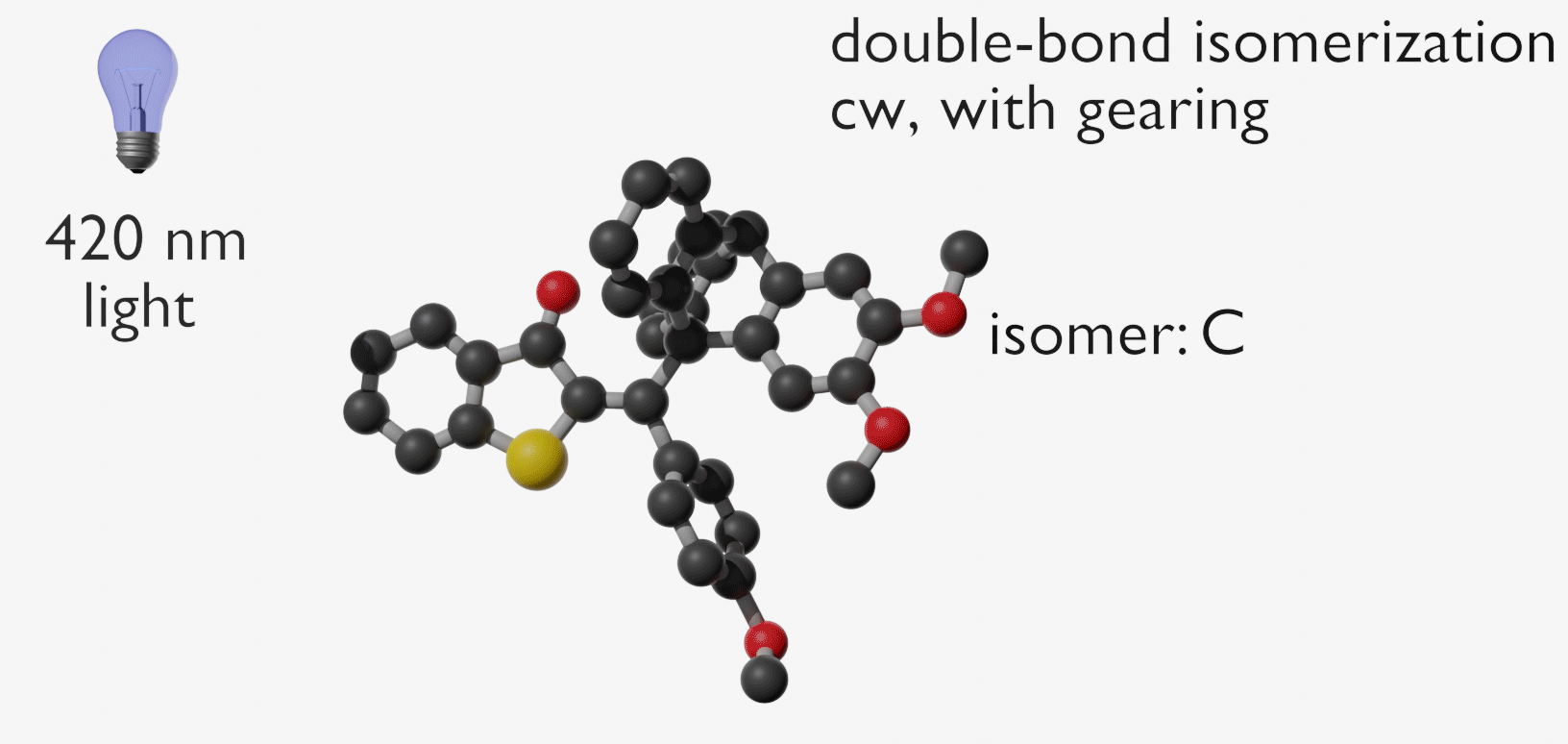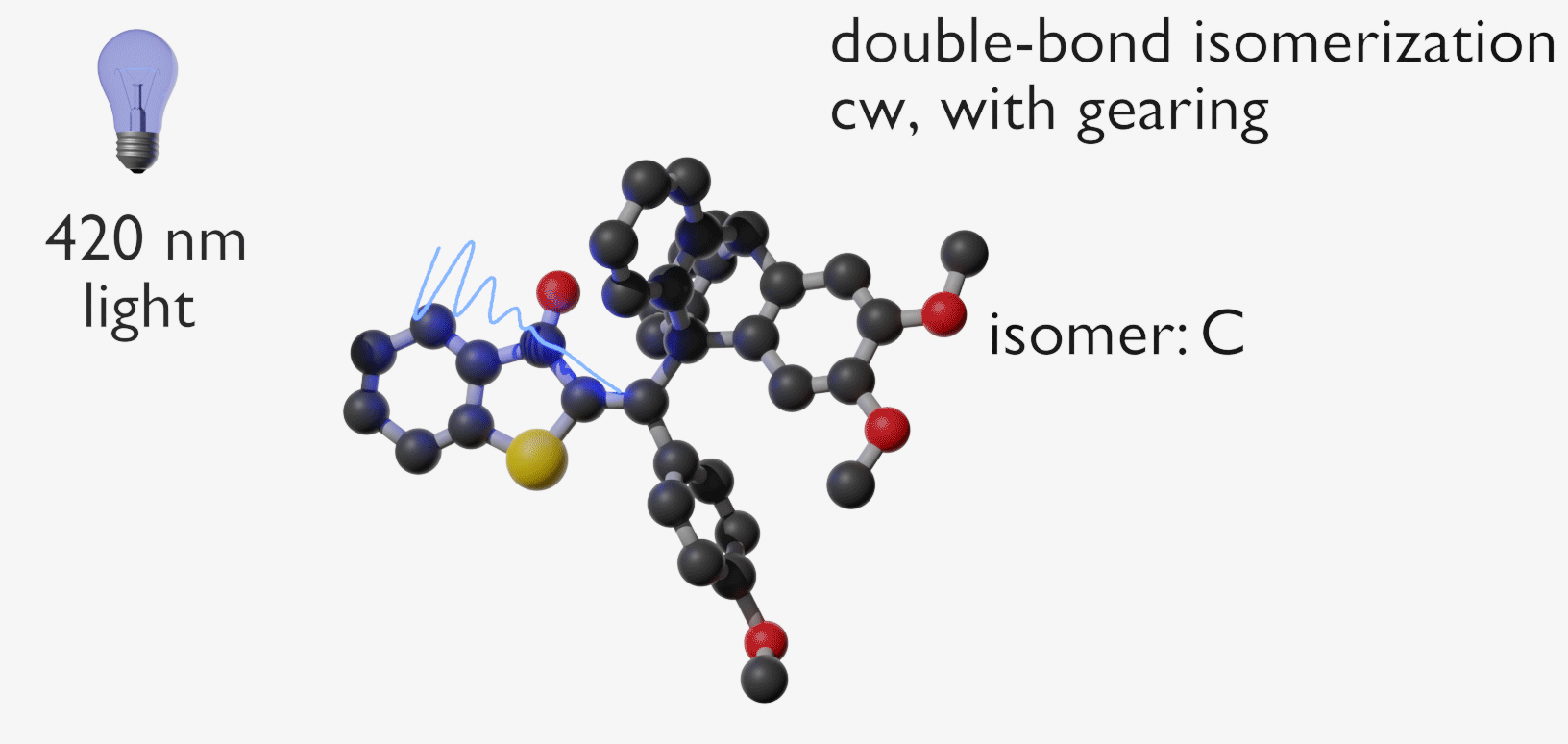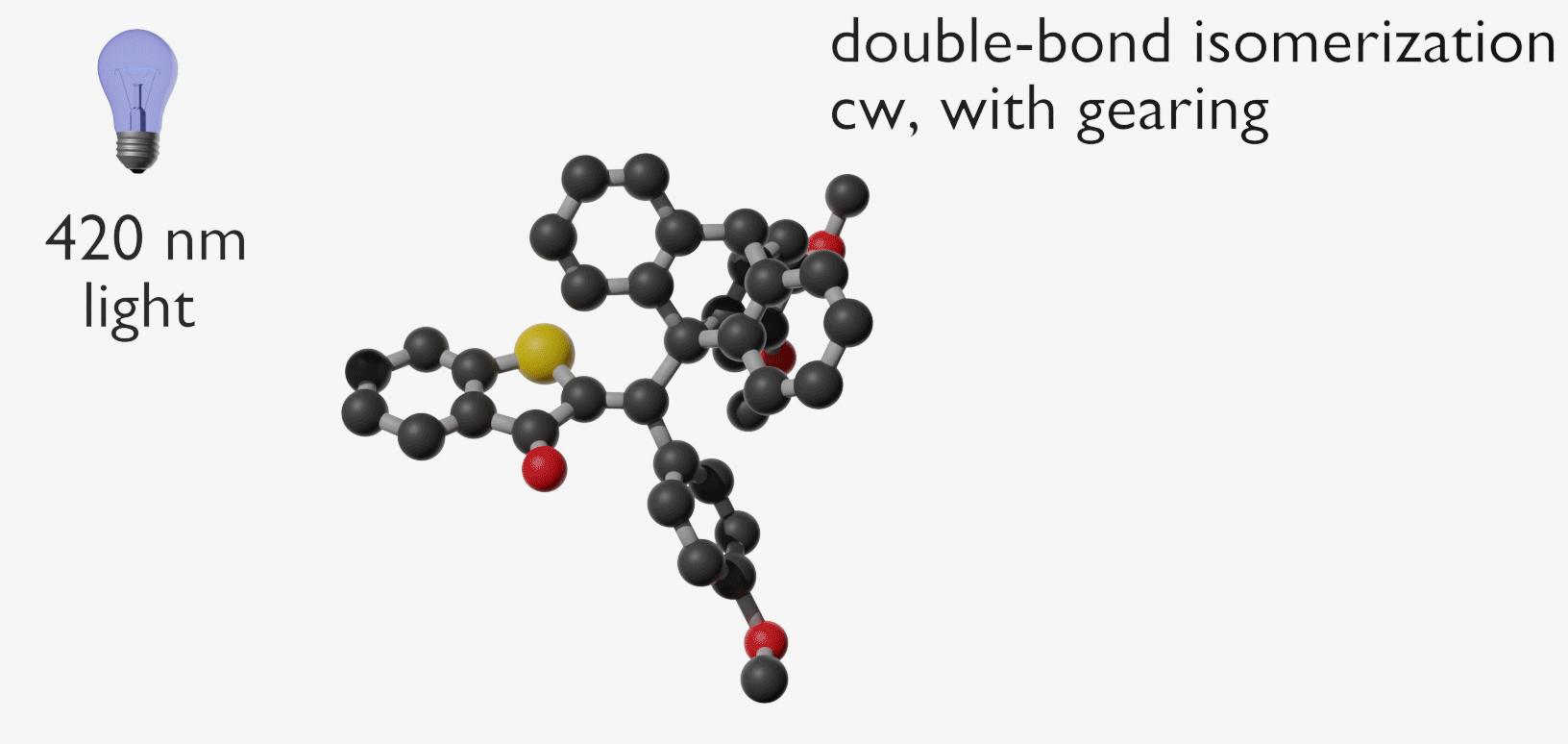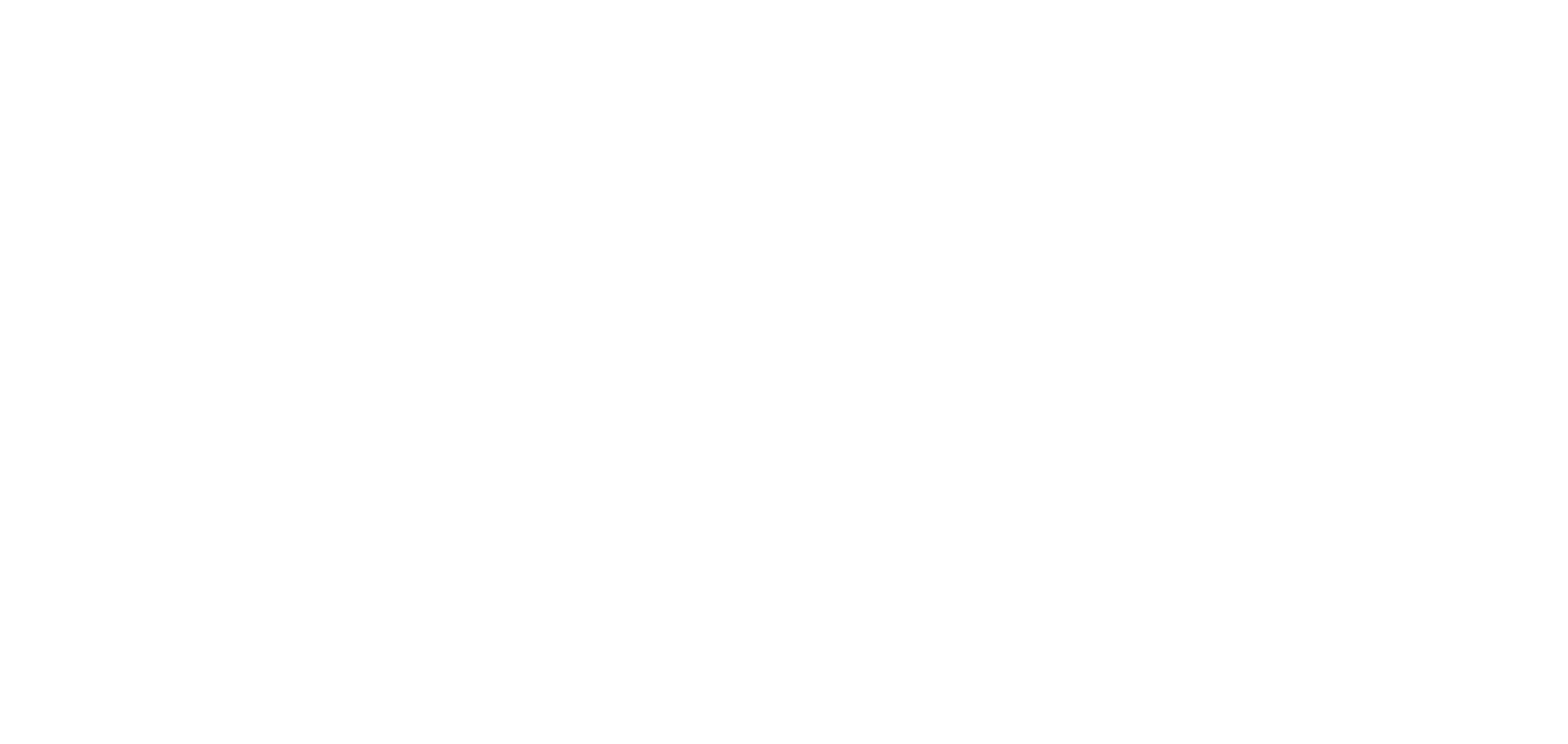
Molecular gears have been investigated early in molecular machine research and studied extensively for decades. So far molecular gearing systems worked by thermal activation in solution leading to the correlated mechanical movement of intermeshed molecular fragments. Because of the thermal nature of this process no directional movements are possible and no means are available to actively power molecular gearing movements. We developed the first energy-fueled molecular gearing system using photons for driving the correlated motion.1 Upon blue light irradiation two molecular fragments move together against each other. Thereby the 180° rotation of the central double bond is translated into a 120° rotation of the adjacent single bond. At the same time the rotation axis is shifted by 120° similar to a macroscopic bevel gear.
Functional Supramolecular Systems
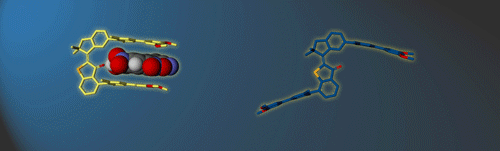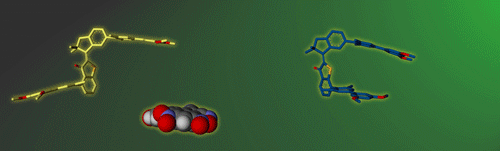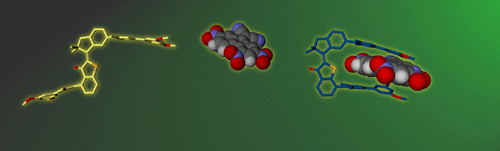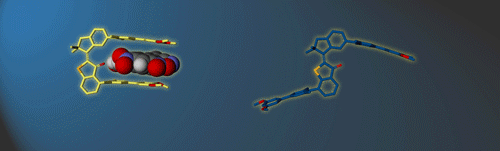
In this research line we develop and study functional supramolecular systems that can be controlled in their properties by external stimuli. We strive to go beyond the sole establishment of molecular recognition processes and implement responsive elements for smart and emerging behavior.
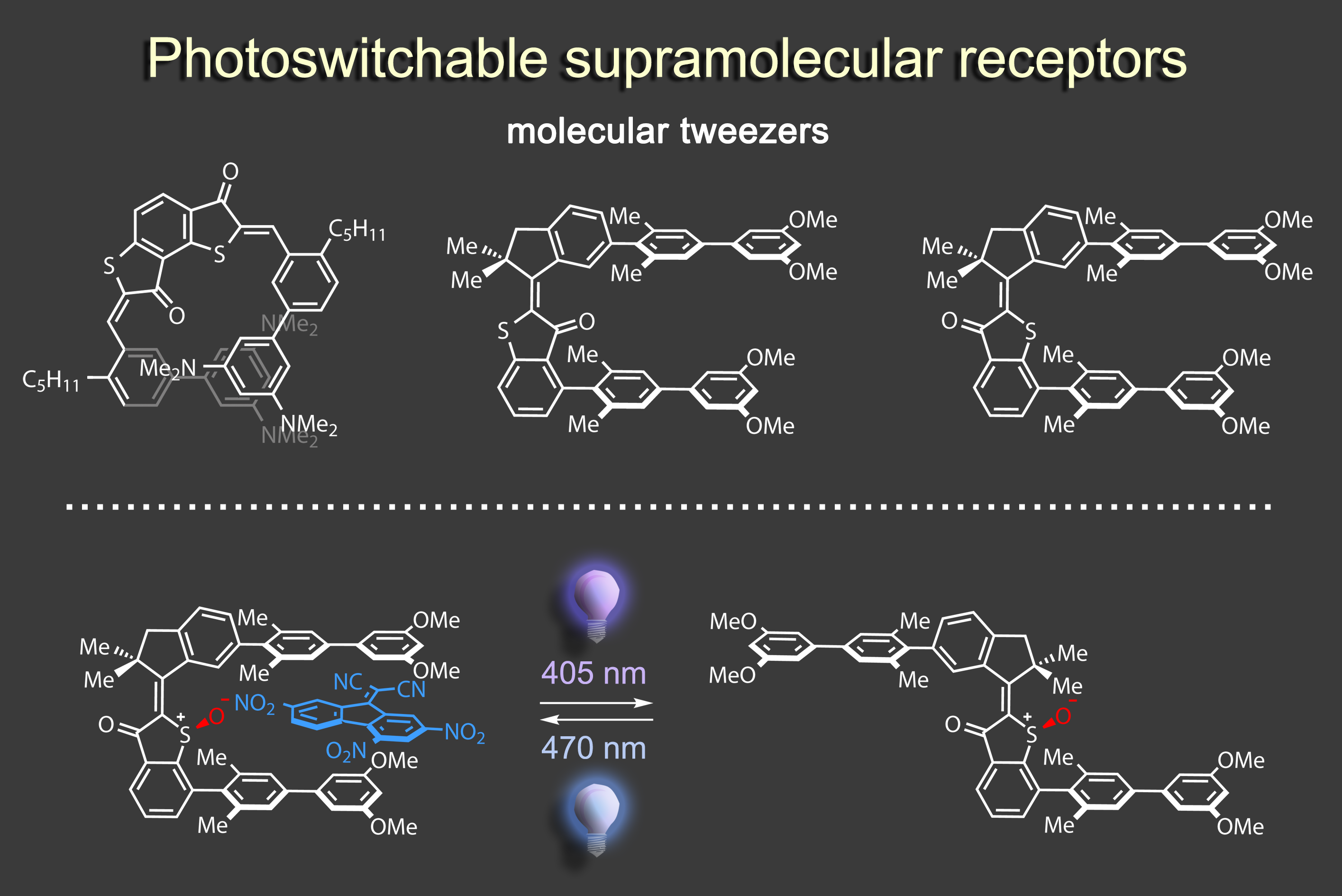
To this end we have created different photoresponsive receptor1 and molecular tweezers motives,2 which we can reversibly switch between high and low affinity states using visible light signals. In a recent effort we were able to elicit a complex and dynamic guest relocation in solution by realizing a new concept: “simultaneous complementary photoswitching”.3 Two complementary substituted molecular tweezers respond to the same wavelength of irradiation in opposite manners. If the first tweezers gain binding affinity the second tweezers lose it at the same time, leading to relocalization of the guest from one host to the other. At a different wavelength of light irradiation the binding affinities and guest residing can be reversed. Only minimal signaling is needed to obtain a multifaceted supramolecular behavior as the result.
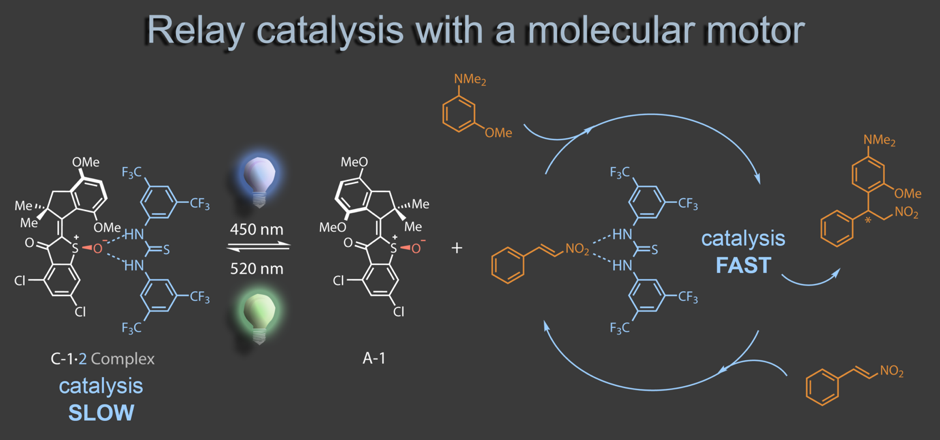
Most recently we started to merge molecular machines with supramolecular chemistry. Using a molecular motor as photoswitchable receptor for hydrogen-bonding organocatalysts the activity of the latter can be made light responsive. In a relay process the organocatalyst can be captured and released from the motor, altering its catalytic activity in a Michael addition reaction. Motor operation thus remote controls catalysis without direct interference.4
- Chem. Eur. J. 2016, 22, 16433.
- Chem. Sci. 2021, 12, 3651.
- Nat. Commun. 2018, 9, 1456.
- J. Am. Chem. Soc. 2020, 142, 19300.
Photochemistry
Photoreactions and Molecular Motions
One core activity in our research concerns chromophore design and mechanistic studies to develop new photoresponsive molecules with unique property profiles. Our long-term goal is gaining absolute control over light-induced molecular motions enabling full spatial and temporal resolution of nano-, micro-, and macroscopic properties.
Focusing on the underexplored class of indigoid photoswitches1 we have established specific molecular designs, which allow us to evoke a range of distinctive bond rotations by irradiation and directly prove them experimentally. Using simple means, like solvent polarity or temperature, different types of such rotations can be interchanged within the same molecule providing exquisite control over multiple molecular motions. Examples are polarity dependent single or double-bond rotation in donor-substituted hemithioindigo2 or the long elusive hula twist, which we evidenced unambiguously in an axially chiral molecular setup.3 This hula twist photoreaction was subsequently studied in a combined effort including ultrafast spectroscopy and excited state theory to gain the first deep insights into its mechanism and the competition with other deexcitation pathways such as TICT formation.4
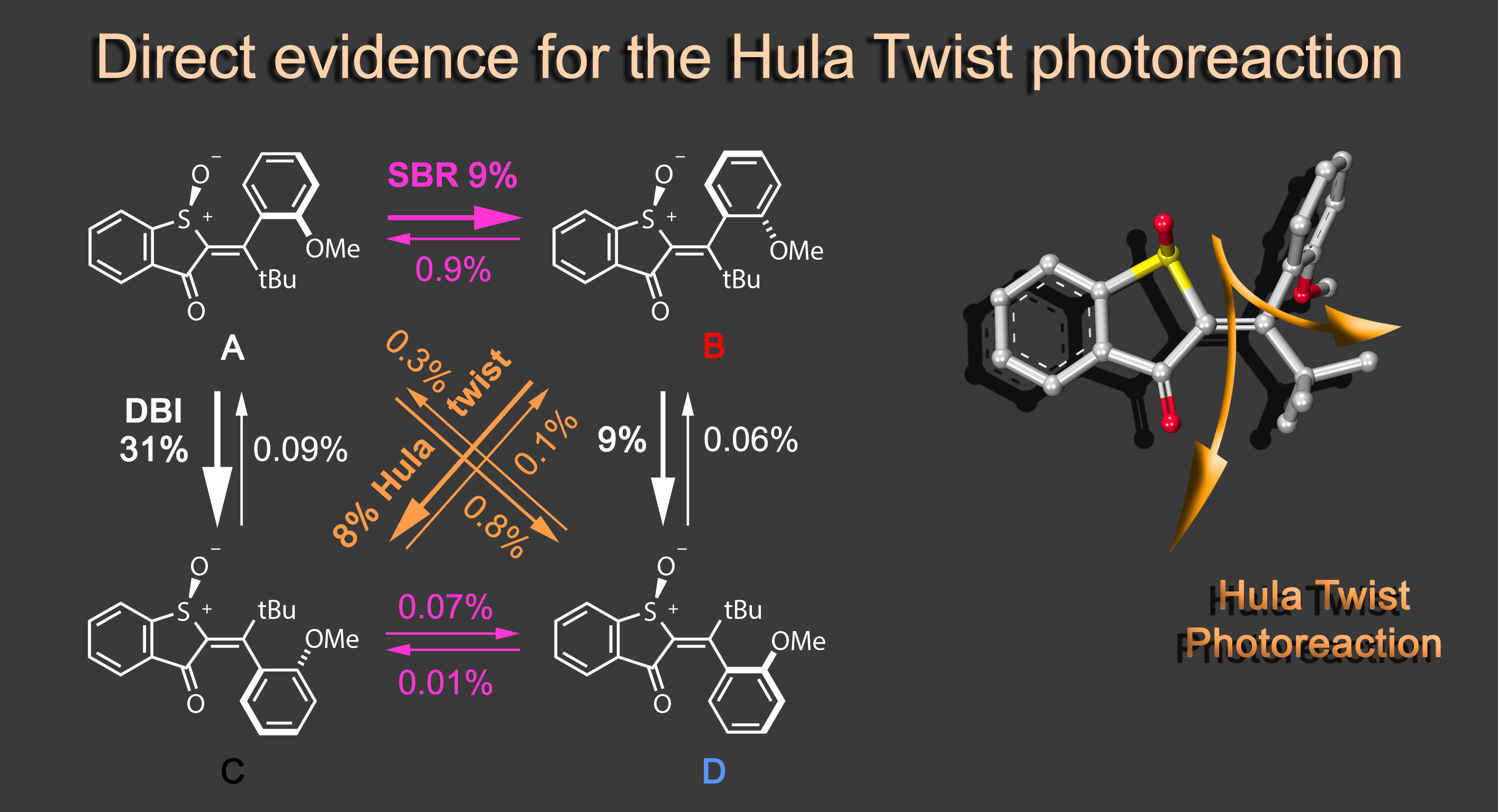
Recently we discovered a hitherto unknown photoreaction – a dual single bond rotation (DSBR) – in which two adjacent single bonds are rotating at the same time upon light irradiation. We use this photoreaction to control the sequential switching of a compact hemithioindigo multi-photoswitch to interchange between a highly selective eight-fold isomer interconversion and a five-fold interconversion. In the different switching sequences hula twist or DSBR photoreactions are followed by thermal single bond rotations (SBR). With this new photoswitching concept an unprecedented density of accessible states and very high control over molecular motions within a simple molecular framework is demonstrated.5
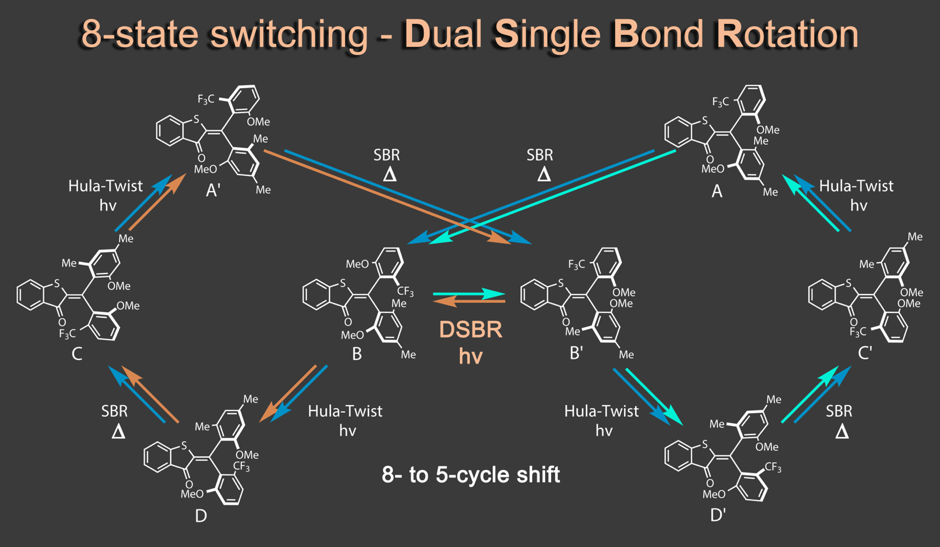
Apart from providing unprecedented insights into fundamental photochemical mechanisms these molecular systems possess especially high potential for the construction of unique future nanomachinery.
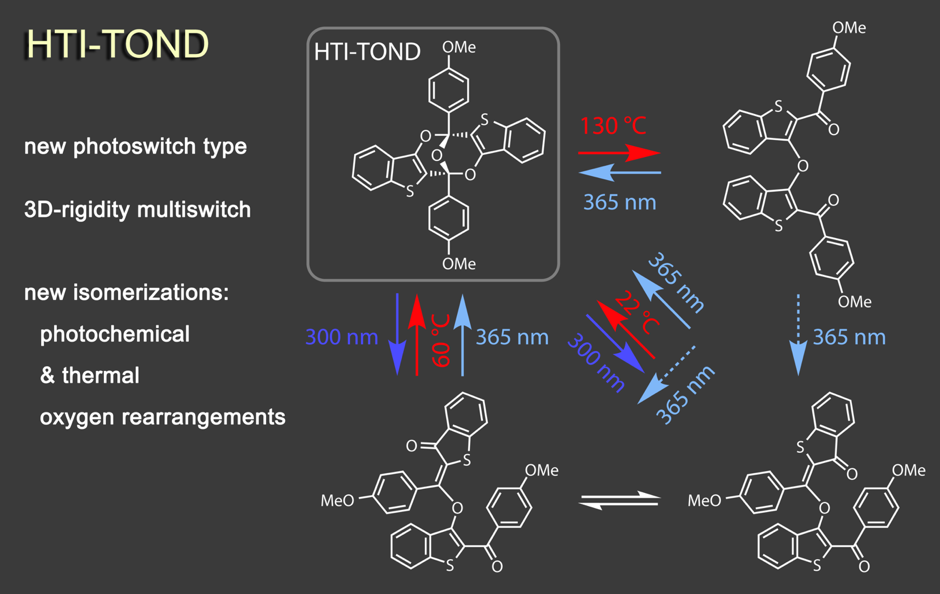
A suite of unique photochemical reactions is found within the trioxobicyclononadiene (TOND) architecture, which we have discovered to be a capable multi-state photoswitch.6 This rigid 3-dimensional structure interconverts with three additional isomers by unusual hetero-Diels Alder photoreactions as well as by hitherto unknown oxygen-rearrangement reactions. TOND photoswitches offer a rare combination of concomitant strong geometric and electronic changes upon switching and an intrinsic four-state nature. They thus provide unique opportunities for the creation of light responsive molecular systems and their applications.
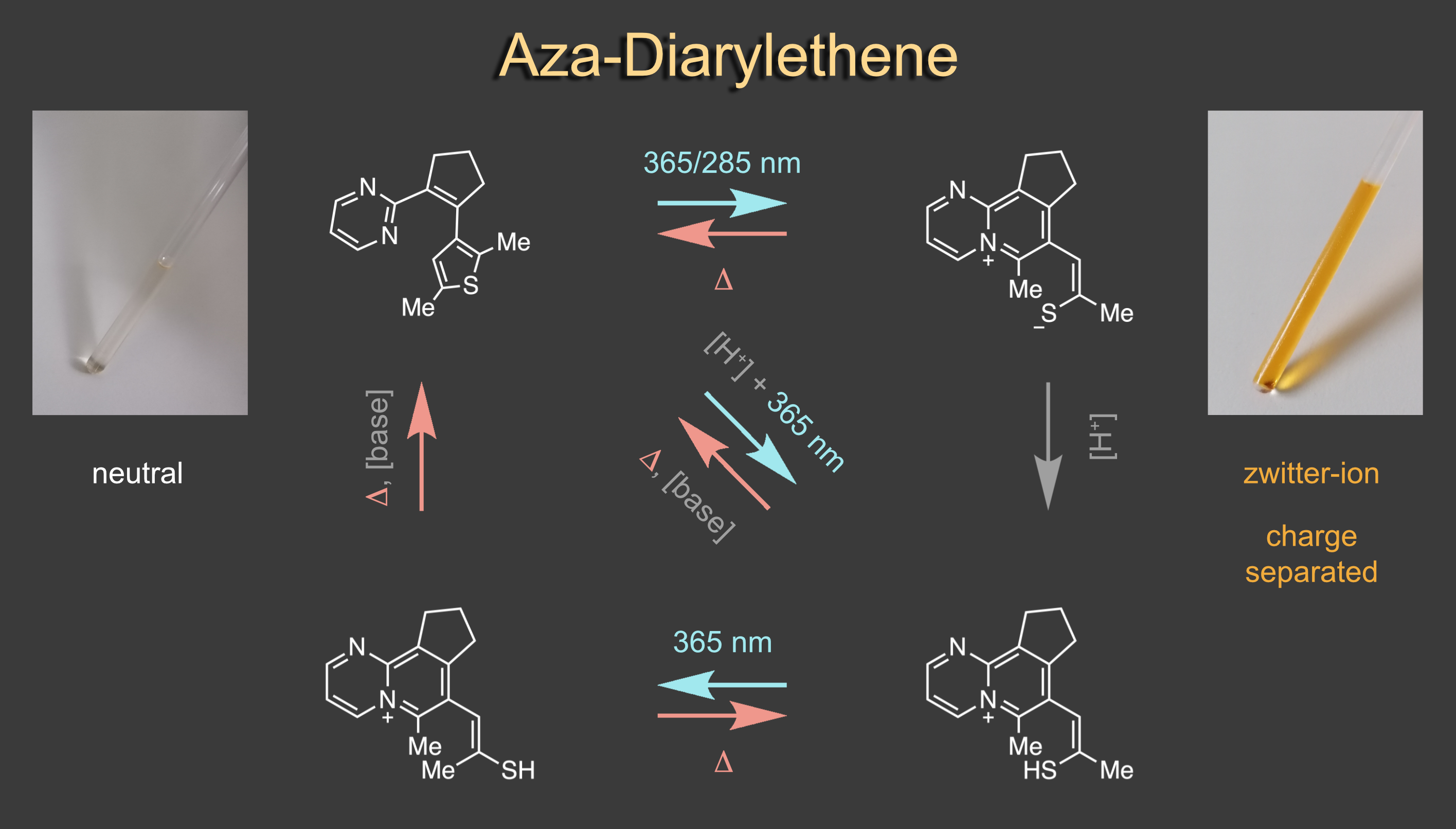
Apart from indigoid chromophores we also scrutinise other types of photoresponsive molecular frameworks. When investigating an aza-diarylethene derivative we discovered a reversible light induced zwitterion formation and concomitant aromatization reaction.7 The zwitterionic product shows negative solvatochromism and reverts completely back to the open aza-diarylethene in a thermally activated process. Thermal stability of the metastable state can be strongly modulated by acid and base additions, allowing to change the T-type photochromism deliberately. With this behaviour aza-diarylethene bridges the behaviours of diarylethenes and merocyanine photoswitches and provides unique photochemical control over charge separation and molecular structure.
- Acc. Chem. Res. 2018, 51, 1153.
- J. Am. Chem. Soc. 2016, 138, 12219.
- Nat. Commun. 2018, 9, 2510.
- J. Am. Chem. Soc. 2023, 145, 14811.
- J. Am. Chem. Soc. 2022, 144, 3029.
- J. Am. Chem. Soc. 2022, 144, 2847.
- J. Am. Chem. Soc. 2024, 146, 9575.
Photoswitches
In a second research line we are developing new types of photoswitches. Of particular importance are highly efficient bistable photoswitches with red light responsiveness, which are interesting for a variety of applications ranging from material sciences to biology and photopharmacology. Hemithioindigo has been the main chromophore structure that we explored for many years, most recently excellent performance was obtained in heterocyclic variants, especially imidazole and indole based derivatives.1 Another capable variant is diaryl-hemithioindigo, which allows additional acid/base switching and applications in advanced molecular logic operations and information processing.2
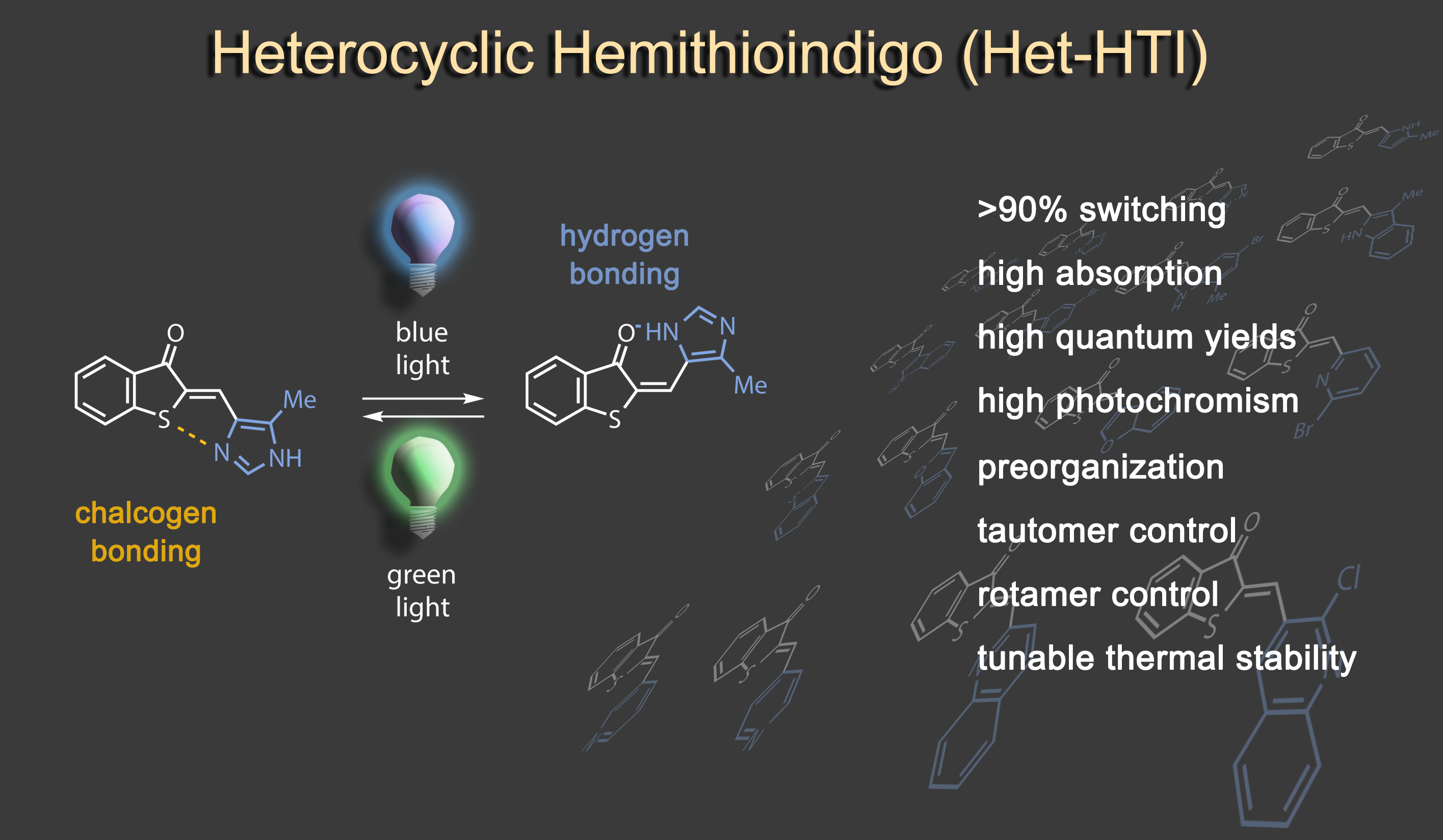
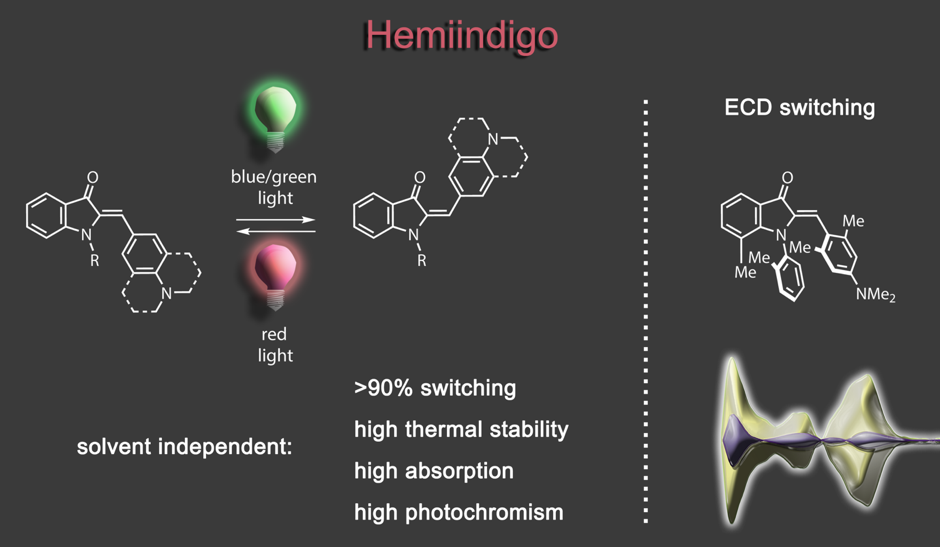
Impressive performances are also given by donor-substituted hemiindigo chromophores allowing photoswitching at the biooptical window with half-lives of the metastable forms reaching >1000 years at ambient temperatures.3,4
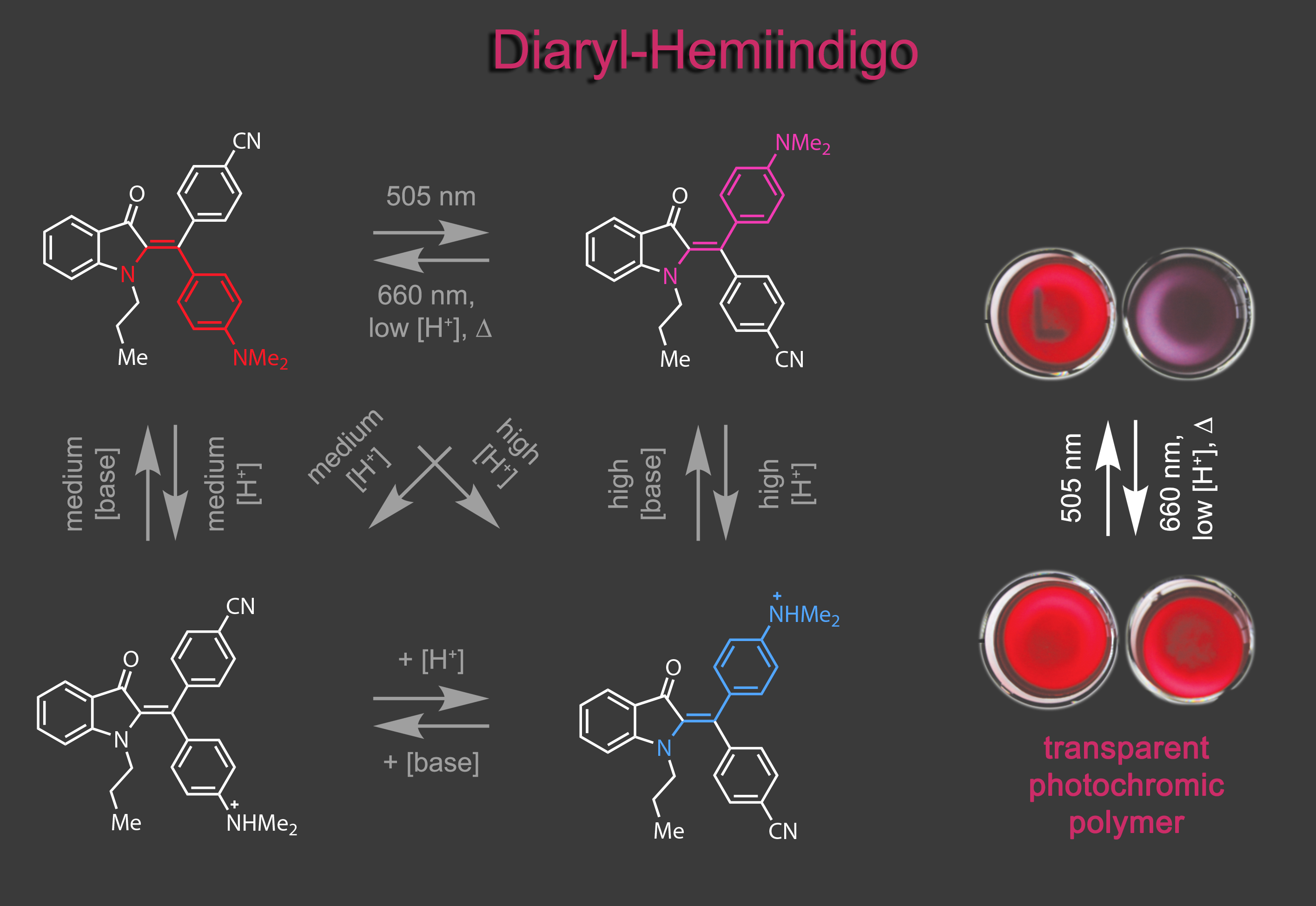
When we gained synthetic access to diaryl-hemiindigo a new switching realm was opened up. Not only are photophysical and photochemical properties improved significantly, but also acid induced pumping of metastable states and acid-gating of photo switching was achieved in a four-state switch. The pronounced color contrast upon photoswitching was harnessed in the first polymer application of our group.5
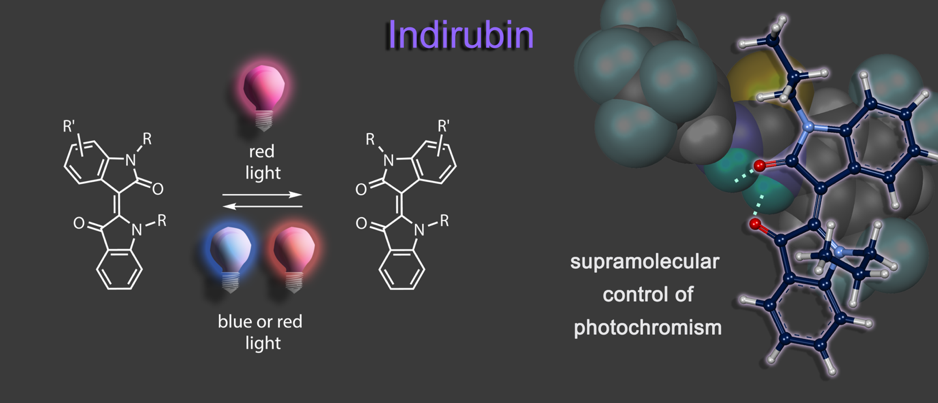
Another addition to molecular photoswitches is the long-known chromophore indirubin, which is rendered into a proficient photoswitch by alkylation. In a supramolecular approach selective hydrogen bonding to the E isomer allows to invert indirubins photochromism and photoswitching in both directions can now be elicited by two different shades of red light.6
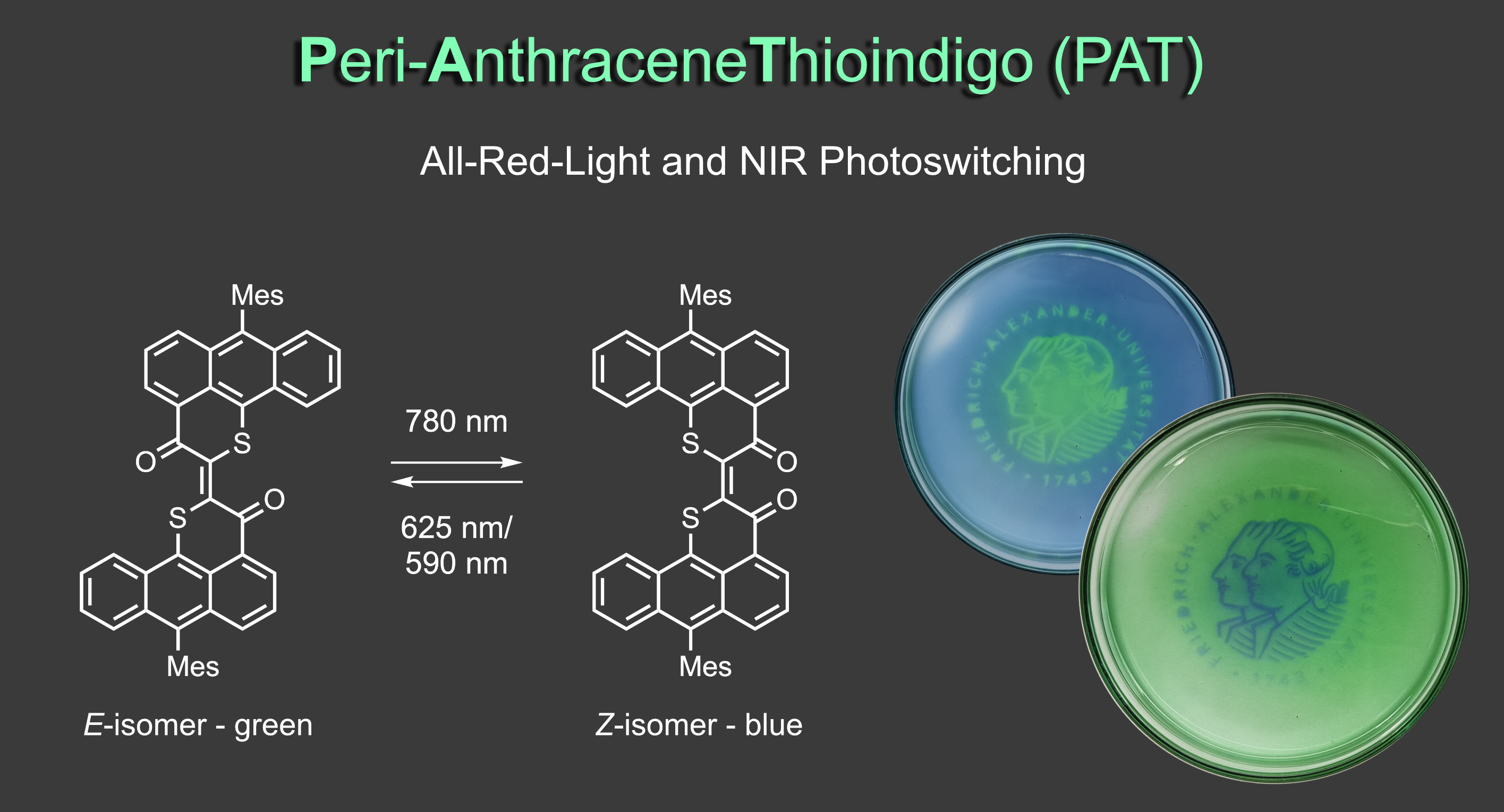
To further redshift the photoresponse to the near infrared (NIR) region of the electromagnetic spectrum and obtain really efficient all-red-light responsiveness we proudly present the Peri-AnthraceneThioindigo (PAT) photoswitch.7 PAT possesses absorptions up to 850 nm, delivers absolutely outstanding all-red-light responsive photoswitching (quantitative E to Z photoisomerization with 780 nm light and 69% or 91% Z to E photoisomerization with 625 nm or 590 nm light, respectively), and is thermally stable for days at 20 °C. It gives high color contrast by changing from bright green to deep blue upon switching and can be applied in photochromic polymers. With these properties PAT pushes the limits of red-light photoswitching and will certainly find many exciting uses further on.
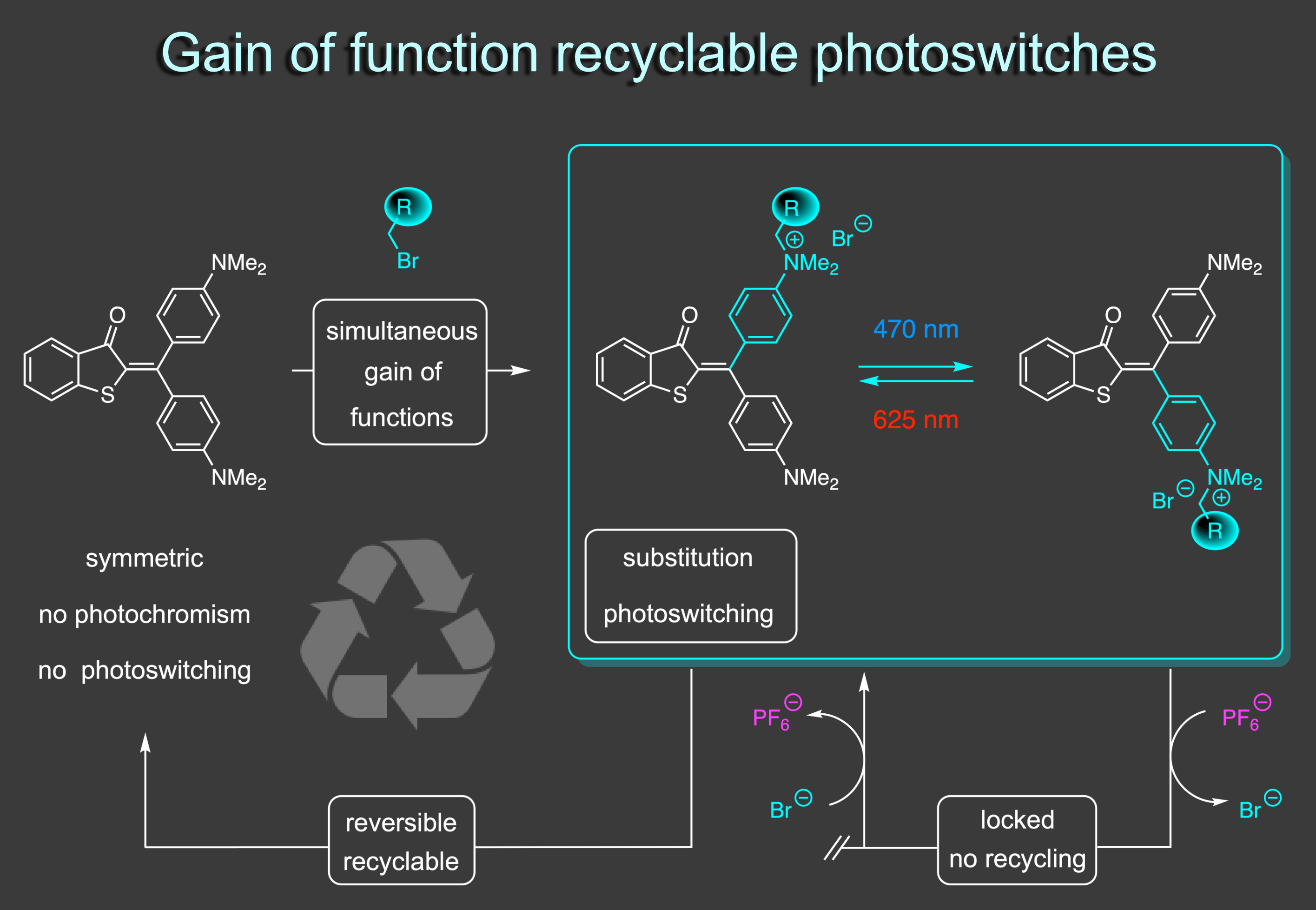
Our recent efforts have led to the development of an entirely new way to think about the generation of photoswitching properties. Usually, photoswitches are substituted for a specific application and then remain dedicated to that task. In our new concept we only generate photoswitching properties when functionalizing a non-photochromic precursor.8 The functionalization reaction simultaneously leads to introduction of a covalently bound group, desymmetriyation of the precursor, and establishment of a strong donor-acceptor cross conjugation. As a result viable photoswitching properties are introduced in response to blue and red light irradiation without background photoreactions of the precursor. The functionalization is fully reversible, which regenerates the symmetric precursor for another functionalization and gain of photoswitching round. In this way recyclable photoswitches are obtained that can be programmed in their properties via the functionalization reaction. Furthermore, recycling can be locked or released by simple anion exchange adding another layer of control to these photoswitches. Exchange to PF6 anions blocks recycling leading to thermally fully stable and functional photoswitches. Reverting to bromide re-enables the recycling again.
- Angew. Chem. Int. Ed. 2022, 134, e202210855.
- Chem. Sci. 2023, 14, 5734.
- J. Am. Chem. Soc. 2017, 139, 15060.
- J. Am. Chem. Soc. 2018, 140, 13558.
- Nat. Commun. 2023, 14, 4382.
- J. Am. Chem. Soc. 2021, 143, 18251.
- Angew. Chem. Int. Ed. 2023, e202312955.
- Angew. Chem. Int. Ed. 2024, e202318767.
Chemical Biology
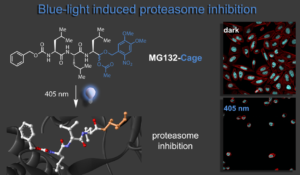
In this research line we develop advanced chemical biology tools for precision regulation of biological processes. Our first approach delivered blue light control over the cell cycle and apoptosis of cancer cells in collaboration with the Zanin lab. To this end we have photocaged the versatile proteasome inhibitor MG132 directly at its reactive aldehyde function keeping the oxidation state unchanged. Upon caging bioactivity is lost and cells proliferate normally. Irradiation releases the inhibitor at a given time and leads to metaphase arrest of the cells. Prolonged exposure causes the apoptosis pathway to be activated and blue light treated cells die. Our light-activated biomolecular tool therefore enables spatial-temporal control of cell fate and was further shown to be compatible with live-cell imaging methods.1
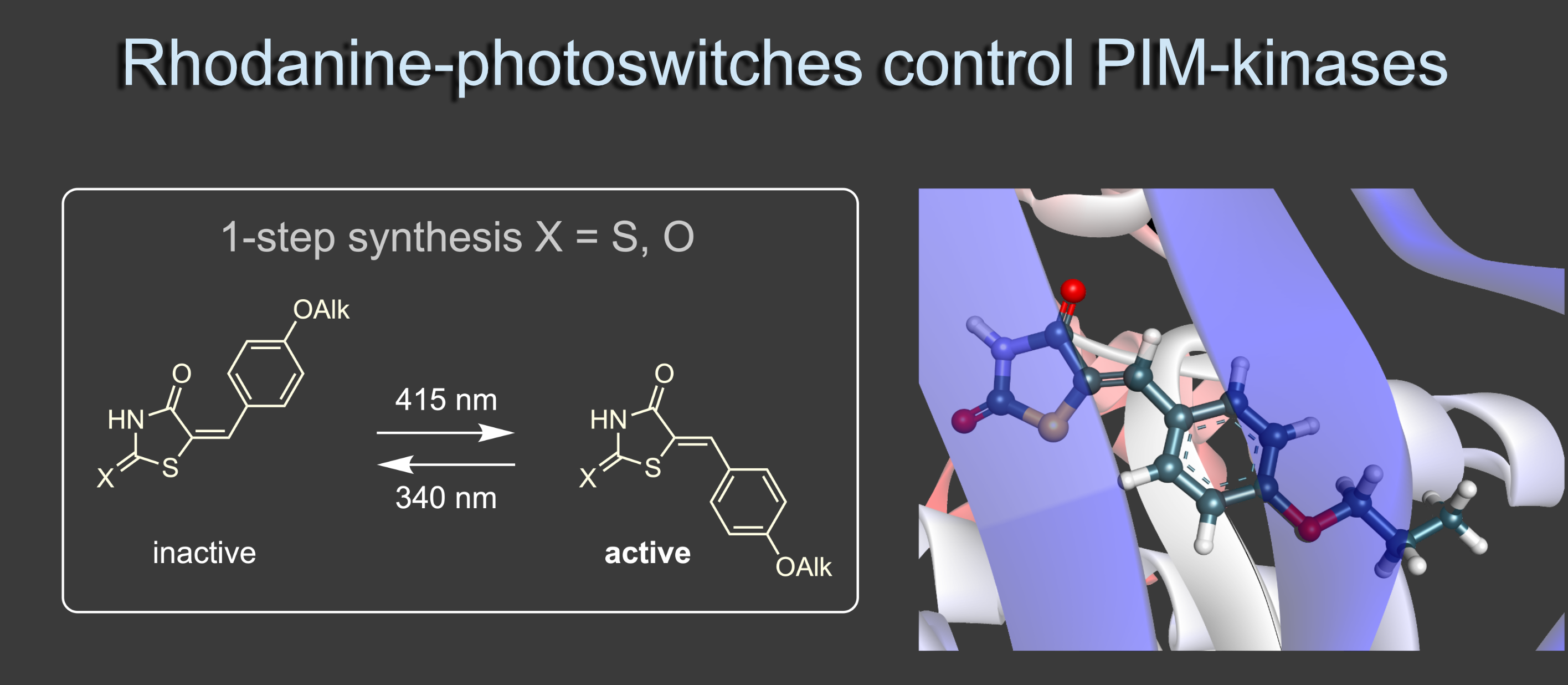
In a second approach we use the reversible photoresponse of indigoid chromophores to modulate inhibition of enzymes vital for cell survival. Rhodanine-based chromophores are ubiquitous molecular structures in different applied fields of chemistry ranging from materials to solar cells and bioactive compounds. We have demonstrated how photoswitching can be elevated to high performance and how rhodanines can be used as advanced photopharmacology tools. In this regard we showed that a known inhibitor of PIM-1 kinase can be strongly photomodulated in its activity allowing for light-controlled apoptosis induction in cancer cells. We also found new derivatives as candidates for potent PIM-1 kinase inhibitors.2