Research
Self-organization is the key building principle of many materials and practically all biosystems. Using molecular simulations, we can understand individual interactions within the context of the co-operative interplay of millions of atoms. This leads to exciting insights into the details of i) the mechanisms of crystal nucleation and growth, but also ii) aspects of molecular life science such as protein folding and biomolecular docking.
Going from understanding to designing, we develop nano-materials that bring together the functionality of molecular recognition and the robustness of atomic packing in inorganic solids. For this endeavor, we closely cooperate with experimental groups and partners from industry in order to transfer findings from molecular dynamics simulations to real-life challenges and their solutions.
Magnetic Water Cleaning
A particulary successful joint venture of experimental characterization of nanomaterials and molecular simulation studies is given by the development of superparamagnetic iron oxide nanoparticles with tailor-made functionality for binding pollutants. Once the nanoparticles are loaded by pollutants, magnetic separation devices can remove the carriers from water – a cleaning technique which is currently develloped to industry-scale.
For example, the designing of hydrophobic surfaces enables the iron oxide particles to bind to nanoplastic particles or to induce the segregation of oil from water (see picture below). Current efforts of molecular modelling aim at increasingly specific use of molecular recognition motifs in order to improve the efficiency of pollutant recognition and to widen the portfolio of surface-functionalization for water remediation from pollution by herbicides, hormones and heavy metals.
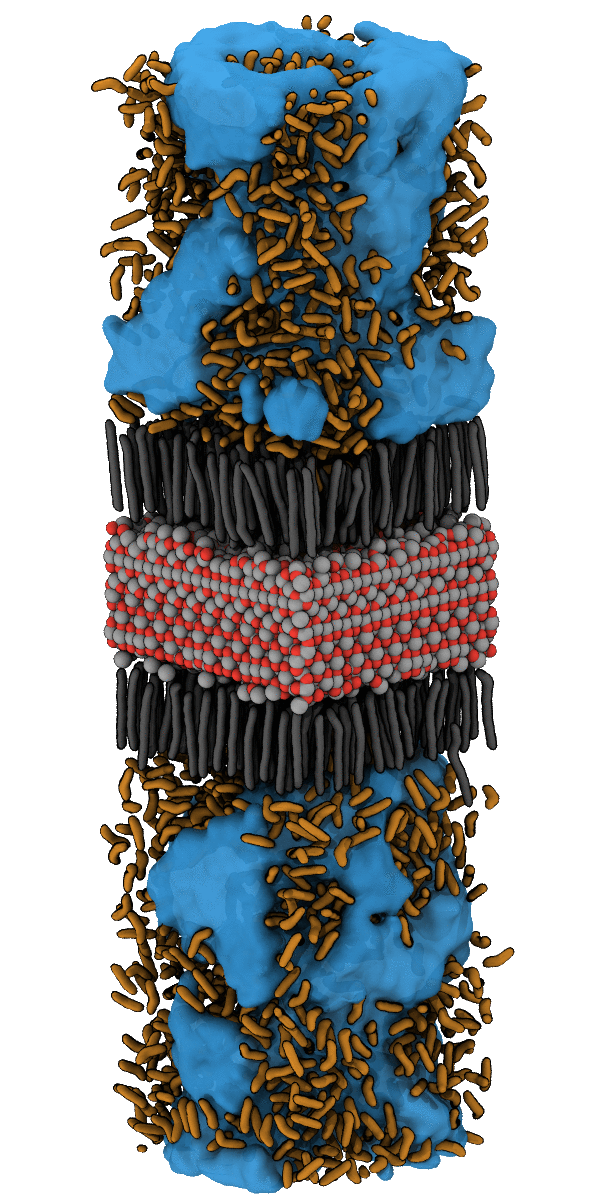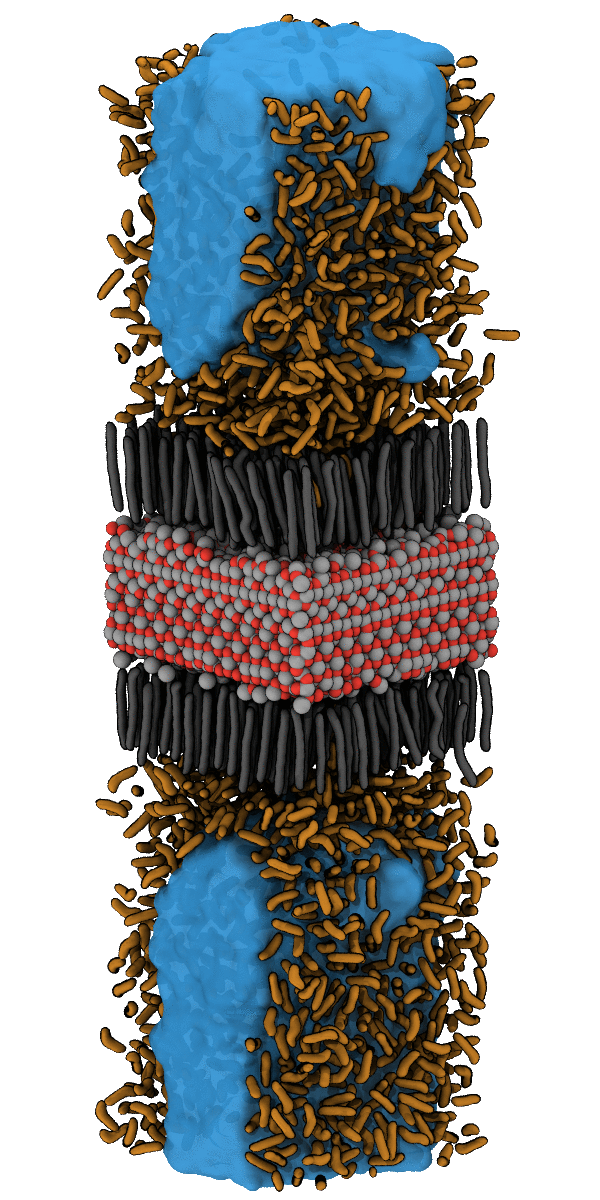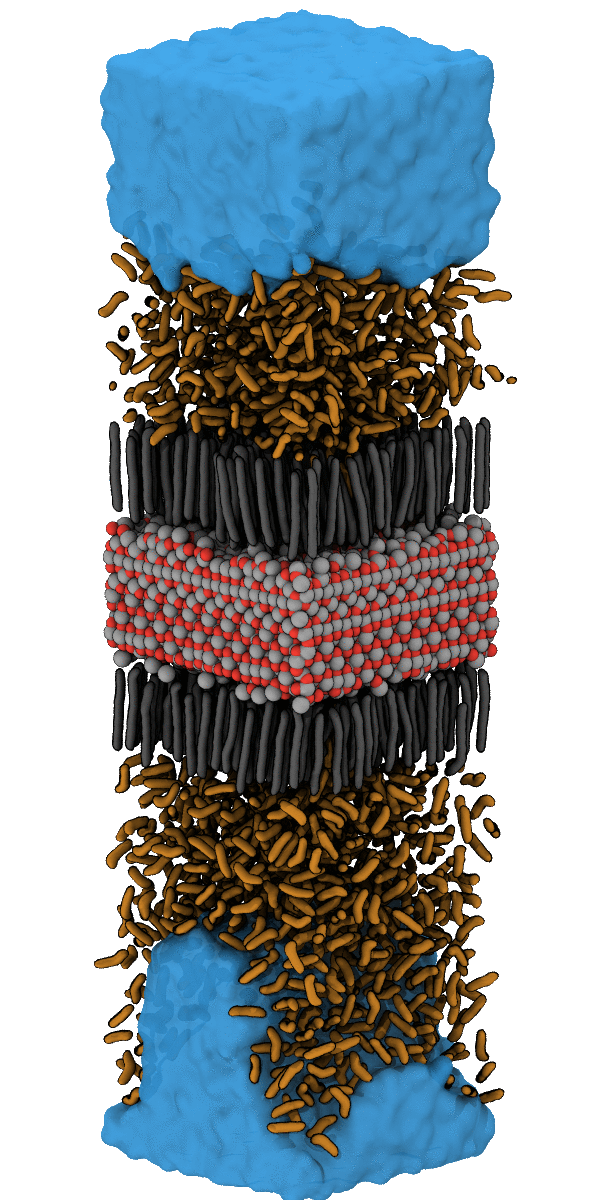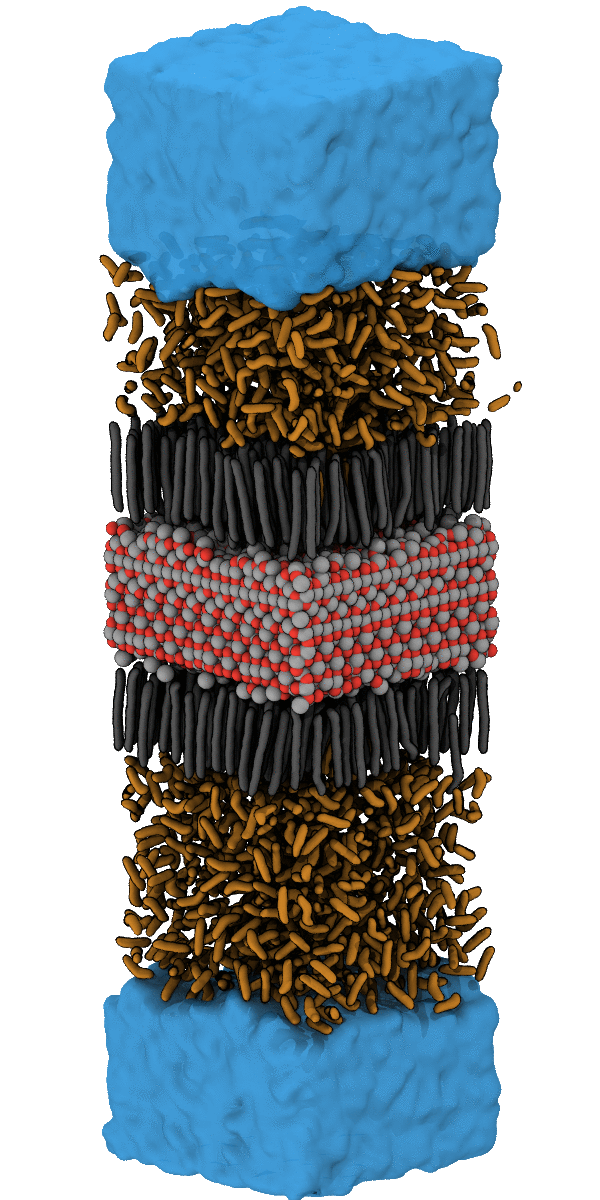
Simulation of the accumulation of n-Heptane on the Nanoparticle SurfaceSimulation of the Accumulation of n-Heptane on the Nanoparticle Surface – see also Halik group for oil-catch in real-life application. |
|
Self-Organization of Crystals, Macromolecules and Composites
Starting from the association of single ions our simulations allow the investigation of the infancy of crystal nucleation from solution. This provides a unique level of insights into (self)-organization and its interplay with ripening reactions and interactions to growth-controlling molecules. Using quantum/classical molecular dynamics simulations, a special focus is dedicated to proton transfer reactions occurring during crystal aggregation and ripening. Likewise, our ‘local pK’ concept enables the assessment of pH-driven crystal dissociation / solution stabilization.
In co-operation with pharmaceutical industry, we explore the solubility of drug molecules for various polymorphs, solvents, pH and thermodynamic conditions (see image below). Likewise, we investigate the stabilization of ionic solutions and the hindering of calcium carbonate precipitation in household devices with partners from consumer products industry.

Nucleation of a molecular crystal from solution: atom scale insights of the infancy of the forming aggregate are used for tailoring preferential crystal motifs to enable polymorph control . |
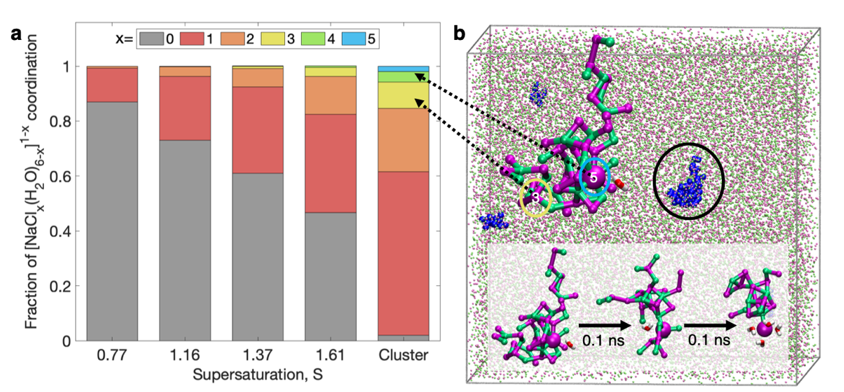
Characterization of ionic solutions (here NaCl in water) at different levels of super-saturization S. Before crystal nucleation, nm sized assemblies of dynamically ordered solvated solutes are observed in both experiment (a) and molecular dynamics simulation (b). |
Materials Simulations
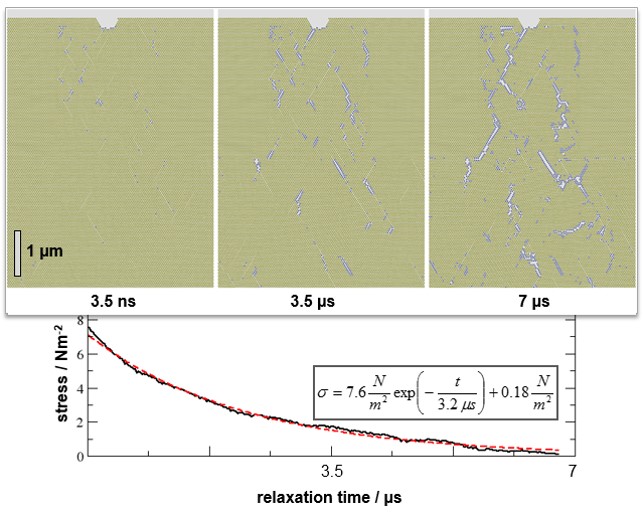
Atomistic and coarse-grained models are used to explore the structure and mechanical properties of nanocrystals and bulk materials. These studies include atomic mobility, defect arrangements, dislocations, grain and phase boundaries as well as their role during deformation and fracture.
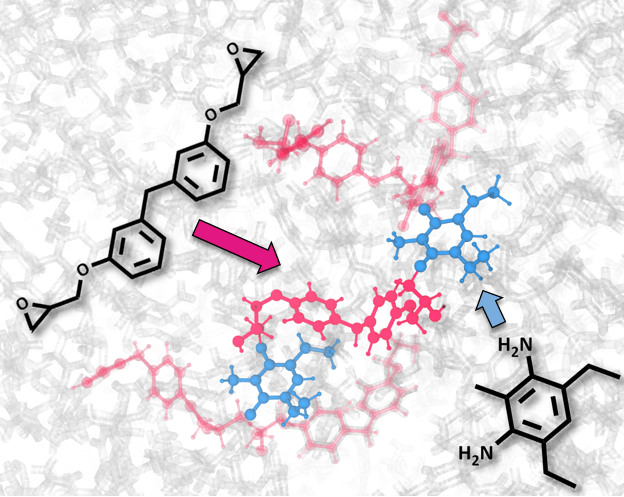
Condensation reactions of epoxy molecules and curing of 3D networks of covalently bonded resins. |
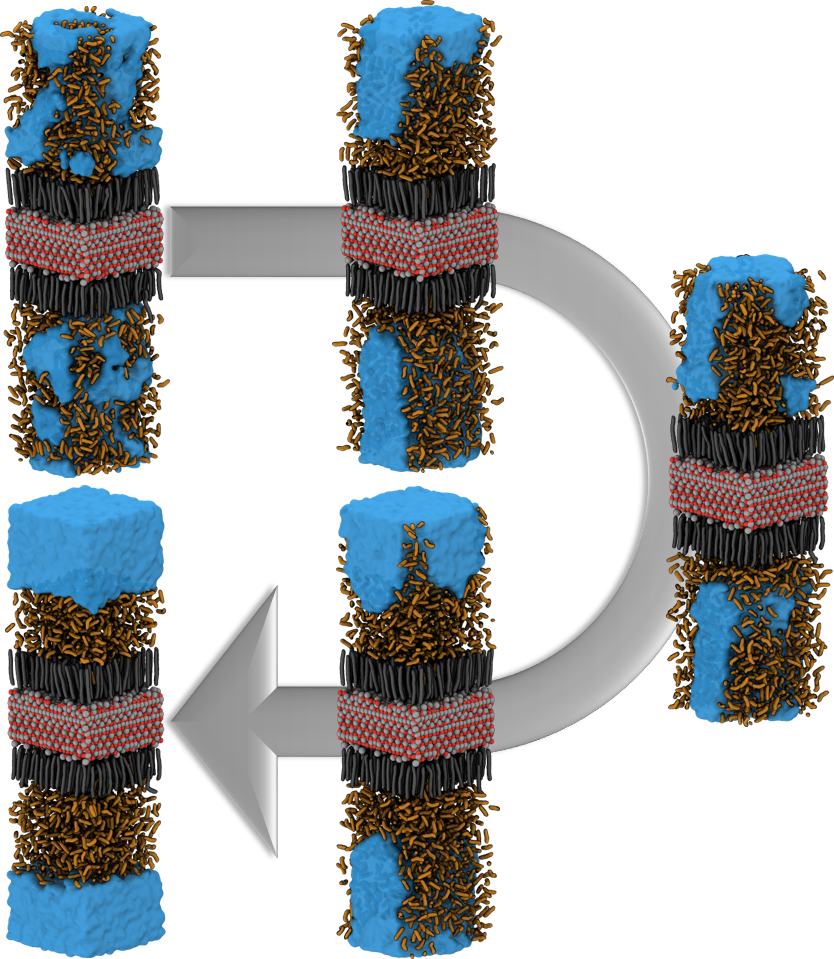
Coarse-grained model of dental enamel and investigation of micro-crack formation upon mechanical load – rationalizing damage control and self-healing of the biomineral. |
Method Development
The investigation of rare barrier crossing events (reactions, nucleation processes) requires efficient simulation strategies. To tackle the time/length scale probleme we apply transition path sampling, constraint MD simulations and related approaches. Diffusion controlled processes, like crystal aggregation from dillute solutions impose different challenges to computer simulation. For this purpose, we devellop specific approaches which allow to study aggregation and growth at the atomistic level of detail.
Force-Field Development
To enable molecular mechanics or efficient QM/MM modelling at high accuracy we develop tailor-made force-fields to extend the scope of off-the-shelf models where needed. Examples are molecular mechanics models of the different states of photo-switches and molecules before/after (de)protonation reactions. Moreover, for the prospering field of modelling additives in oil and at oil-steel interfaces, our OilF force-field is continuously extended to enable the molecular simulation of anti-wear films, debris dispergion and tribology at the nm scale.

Molecular dynamics simulation of a lubricated contact of two machine parts. While the base oil is weakly attached to either of the solid surfaces, suitable additives tend to remain and act as friction modifiers. |
