Research
We are actively involved in EXC35 Engineering of Advanced Materials, SFB953 Synthetic Carbon Allotropes, and GRK1910 Medicinal Chemistry of Selective GPCR Ligands.
Semiempirical MO-Theory
The older semiempirical molecular-orbital program VAMP and its successor EMPIRE were both written in the group and EMPIRE is the subject of continued development. EMPIRE [1] was designed from scratch as a highly parallel, high-performance program for NDDO-based semiempirical MO calculations on very large systems. It has been tested up to 100,000 atoms on 1,024 processors. The current version of EMPIRE can perform single-point calculations and geometry optimizations using RHF or UHF formalisms and for both molecular and 1D- 2D- and 3D-periodic systems. [2] It performs well in parallel on both multi-core workstations and on clusters. EMPIRE calculations can be run from a completely web-based user-interface and the results visualized. The single-node (workstation) version of EMPIRE is available free to bona fide academic users.
The following Figure shows a small EMPIRE calculation of a single hydrogen-terminated graphene sheet. The molecular electrostatic potential is projected as color coding onto the standard isodensity surface.
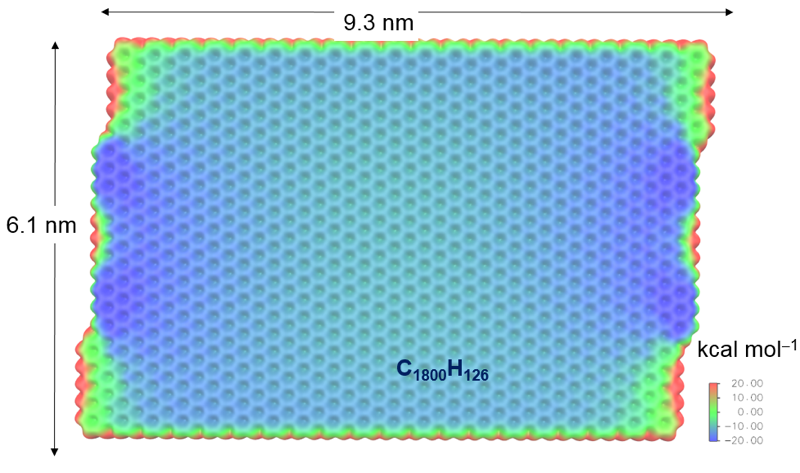
The group has also developed and parameterized the semiempirical Hamiltonian AM1*, which is based on AM1 for the elements H, C, N, O and F and includes d-orbitals fo second-row elements and transition metals. [3], [4], [5], [6], [7], [8], [9], [10], [11]
Modeling Soft Electronic Devices
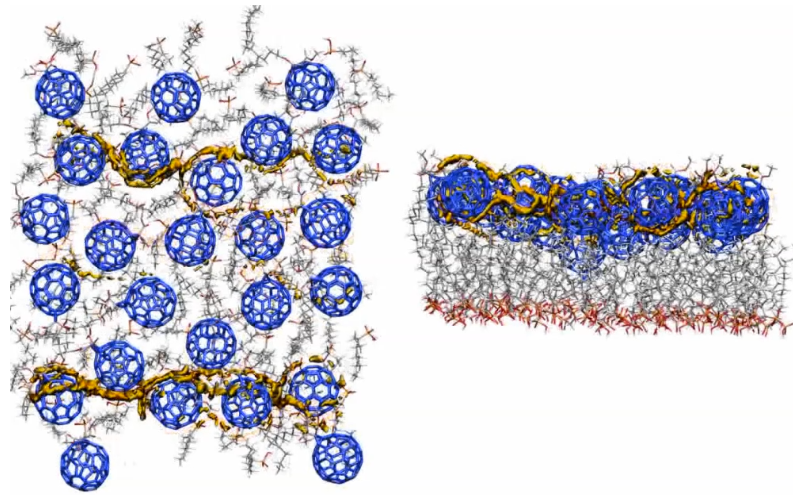
We pioneered the technique of using the local ionization energy and local electron affinity [12] as external potentials for simulations of charge-carrier movement in organic semiconductors or devices. This concept started with simple visualization of charge-transfer paths [13] and was extended to Monte Carlo searches of the conduction paths themselves, [14] as shown in the Figure below for a self-assembled monolayer field-effect transistor (SAMFET). The left-hand picture shows the SAM from above and the right-hand one from the side. The conduction paths through the fullerene n-type semiconductor channel can be seen clearly. This work has been reviewed in the open-access literature.
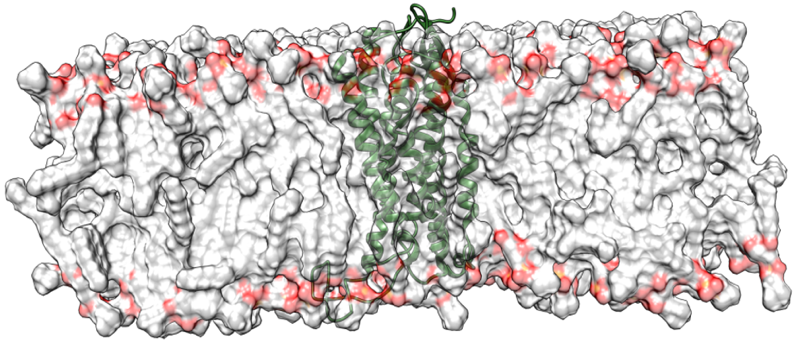
Long-term molecular-dynamics simulations on switchable protein systems
We initially investigated the tetracycline repressor, TetR as an example of a signal-transduction protein that is switched by a small molecule. [15] This work suggests that X-ray structures of such proteins do not necessarily reflect whether the protein is induced or not, but that long-term molecular-dynamics simulations can identify the induction state reliably. More recently, we have worked on the nuclear receptor hNur77 and a variety of G-protein-coupled receptors (GPCRs) within GRK1910 Medicinal Chemistry of Selective GPCR ligands. This work involves enhanced-sampling techniques applied to ternary GPCR-ligand-G-protein complexes in order to identify binding sites and mechanisms.
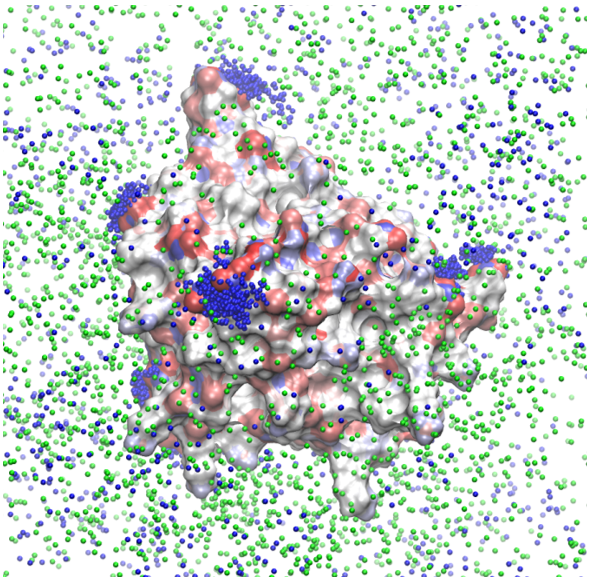
Specific-ion effects in aggregation phenomena
We have used molecular-dynamics simulations to determine the effect of counterions on the aggregation of polycarboxylate structurally conserved micelles. Sodium counterions stabilize the micelles by forming bridged contact ion pairs at the surface of the micelle. The figure below shows the density of sodium ions around a micelle:

The same effects are also observed for aggregating proteins, for instance β-lactoglobulin. [16] The sodium counterions are shown in blue in the snapshot below:
Non-covalent interactions
Our work on non-covalent interactions has ranged from our seminal contributions to the concept of σ-hole bonding, [17], [18], [19] which includes hydrogen bonding. The figure below shows the molecular electrostatic potential at the standard isodensity surface of chlorobenzene; the σ-hole can be seen clearly:

However, dispersion interactions can also be significantly anisotropic, as shown for the interaction of an argon atom with bromine: [20]

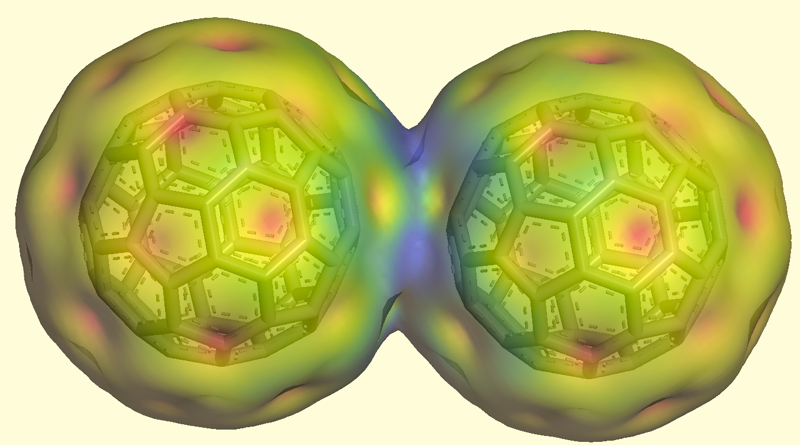
More recently, we discovered the unusual inter-fullerene one-electron bond: [21]
-
EMPIRE: A highly parallel semiempirical molecular orbital program: 1: Self-Consistent Field Calculations, M. Hennemann and T. Clark, J. Mol. Model., 2014, 20, 2331 (DOI: 10.1007/s00894-014-2331-4).
-
EMPIRE: A highly parallel semiempirical molecular orbital program: 2: Periodic boundary conditions, J. T. Margraf, M. Hennemann, B. Meyer and T. Clark, J. Mol. Model., 2015, 21, 144 (DOI: 10.1007/s00894-015-2692-3)
-
AM1* Parameters for Phosphorous, Sulfur and Chlorine, P. Winget, A. H. C. Horn, C. Selçuki, B. Martin, and T. Clark, J. Mol. Model., 2003, 9, 408-414 (DOI: 10.1007/s00894-003-0156-7).
-
AM1* Parameters for Aluminum, Silicon, Titanium and Zirconium, P. Winget and T. Clark, J. Mol. Model., 2005, 11, 439−456 (DOI: 10.1007/s00894-005-0236-y).
-
AM1* Parameters for copper and zinc, H. Kayi and T. Clark, J. Mol. Model., 2007, 13, 965-979 (DOI: 10.1007/s00894-007-0214-7).
-
AM1* parameters for bromine and iodine, H. Kayi and T. Clark, J. Mol. Model., 2009, 15, 295-308 (DOI: 10.1007/s00894-008-0419-4).
-
AM1* parameters for vanadium and chromium, H. Kayi and T. Clark, J. Mol. Model., 2009, 15, 1253-1269 (DOI: 10.1007/s00894-009-0489-y).
-
AM1* parameters for cobalt and nickel, H. Kayi and T. Clark, J. Mol. Model., 2010, 16, 29-47 (DOI: 10.1007/s00894-009-0503-4).
-
AM1* parameters for manganese and iron, H. Kayi and T. Clark, J. Mol. Model., 2010, 16, 1109-1126 (DOI: 10.1007/s00894-009-0614-y).
-
AM1* parameters for palladium and silver, H. Kayi and T. Clark, J. Mol. Model., 2011, 17, 2585-2600 (DOI: 10.1007/s00894-010-0940-0).
-
AM1* parameters for gold, H. Kayi, J. Mol. Model., 2010, 16, 1029-1038 (DOI: 10.1007/s00894-009-0613-z).
-
Local molecular properties and their use in predicting reactivity, B. Ehresmann, B. Martin, A. H. C. Horn and T. Clark, J. Mol. Model., 2003, 9, 342-347 (DOI: 10.1007/s00894-003-0153-x).
-
Tuning electron transfer through p-phenyleneethylene molecular wires, C. Atienza, N. Martin, M. Wielopolski, N. Haworth, T. Clark and D. Guldi, J. Chem. Soc. Chem. Commun., 2006, 3202-3204. (DOI: 10.1039/b603149h).
-
Improving the Charge Transport in Self-assembled Monolayer Field-effect Transistors – From Theory to Devices, C. M. Jäger, T. Schmaltz, M. Novak, A. Khassanov, A. Vorobiev, M. Hennemann, A. Krause, H. Dietrich, D. Zahn, A. Hirsch, M. Halik and T. Clark, J. Am. Chem. Soc., 2013, 135, 4893-4900 (DOI: 10.1021/ja401320n).
-
Induction of the Tetracycline Repressor: Characterization by Molecular-Dynamics Simulations, F. Haberl, H. Lanig and T. Clark, PROTEINS: Structure, Function, and Bioinformatics, 2009, 77, 857-866 (DOI: 10.1002/prot.22505).
-
Carboxylate Ion-Pairing with Alkali-Metal Ions for β-Lactoglobulin and its Role on Aggregation and Interfacial Adsorption, F. R. Beierlein, T. Clark, B. Braunschweig, K. Engelhardt, L. Glas and W. Peukert, J. Phys. Chem. B, 2015, 119, 5505-5517 (DOI: 10.1021/acs.jpcb.5b01944).
-
Halogen Bonding: The σ-Hole, T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model. , 2007, 13, 291-296 (DOI: 10.1007/s00894-006-0130-2).
-
Directional Weak Interactions: σ-Hole Bonding, J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aus. J. Chem, 2010, 63, 1598-1607 (DOI:10.1071/CH10259).
-
σ-Holes, T. Clark, WIREs Comput. Mol. Sci., 2013, 3, 13-20 (DOI: 10.1002/wcms.1113).
-
Directional Non-covalent Interactions: Repulsion and Dispersion, A. El Kerdawy, J. S. Murray, P. Politzer, P.Bleiziffer, A. Hesselmann, A. Görling and T. Clark, J. Chem. Theor. Comput., 2013, 9, 2264–2275 (DOI: 10.1021/ct400185f).
-
Fullerene van der Waals Oligomers as Electron Traps, T. E. Shubina, D. I. Sharapa, C. Schubert, D. Zahn, M. Halik, P. A. Keller, S. G. Pyne, S. Jennepalli, D. M. Guldi and T. Clark, J. Am. Chem. Soc., 2014, 136, 10890–10893 (DOI: 10.1021/ja505949m).